Scientific Programming¶
This course is brought to you by:
-
e-mail: teso _AT_ disi _DOT_ unitn _DOT_ it
Office hours: Tuesday, 14:00-15:00, Room 143 Povo 2.
-
e-mail: t _DOT_ tebaldi _AT_ unitn _DOT_ it
Remaining classes:
- 21/11/2016
- 5/12/2016
Finals will be on:
- 24/1/2017, 9:00-11:00
- 8/2/2017, 14:00-16:00
Syllabus¶
The course will offer a (rather thorough) introduction to the Python language, as well as to Python packages relevant to biological and scientific applications.
We will work with the following Python packages:
- biopython, for managing biological resources
- pandas, for dealing with tabular data, statistics and visualization
- numpy, for general-purpose numerical manipulation
Note
There are many more Python packages out there!
Feel free to browse through PyPI - the Python Package Index for a wide selection of packages. At the time of writing, it hosts 89422 distinct packages!
Bibliography¶
Books:
| [book1] |
|
| [book2] | (1, 2, 3)
|
Other useful material:
- The Python for Biologists website is tailored for biologists.
- Bassi, A Primer on Python for Life Science Researchers, PLoS Computational Biology, 2007.
- The official Python website can be found here. There is also a Getting Started section you may want to peruse.
- A few slides on programming languages and the basics of Python.
Evaluation¶
There will be two midterms, consisting of practical programming tests on the two halves of the program, roughly corresponding to the Python language sans external libraries, and the usage of scientific libraries.
There will be an (alternative) final exam, consisting of a practical programming test.
In both the midterms and the final, the sudent will be asked to write a (short) Python program making use of the technologies presented during the course. The score will depend on the student’s proficiency in Python programming.
Running a Linux Distro¶
In order to run a Linux distribution “inside” your Windows installation, you can use a Virtual Machine (VM). A VM is a “virtual computer running inside your computer”.
There are several VM players available. A few options:
- VirtualBox
- VMware, look for the “VMware player” application
- Parallels
There is also a Free & Open Source alternative, QEMU, but it is rather unix-centric and definitely the least user-friendly of the pack.
Now download a Linux distribution. I suggest downloading a user-friendly distribution, such as:
Download the installation image (also called ISO), it represents a CD-ROM or a DVD-ROM.
Now run the VM player and tell it to create a new virtual machine (details depend on the player you use). Tell it to install the Operating System from the installation image you already downloaded. There are several tutorials on how to do this, like this one.
The Linux installation menu will appear on screen. Just follow the instructions.
Hopefully everything will work out, and in a few minutes Linux will be installed in your Virtual Machine.
If you have trouble, try to ask a more experienced computer user (your colleagues, Toma, or me), or search e.g. “how to install Ubuntu on VirtualBox” on Google. There are a lot of useful tutorial online
Unix shell: a few tips¶
Warning
The material in this section does not play any role in the exam! It is only given for students to get a bit familiar with the Unix shell.
Opening a shell. When you open a new terminal, a new shell prompt is shown:
stefano@computer:~/$
The exact text changes between Unix distributions/versions.
In my case, the login text tells me what is the name of my user on the machine
I am using (stefano), what is the name of the computer I am using
(computer), and in what directory I am standing in (in this case my home
directory ~).
The tilde character ~ is just a shorthand for the full path of my home
directory:
/home/stefano/
Running commands. In order to run a command, say python, it is
sufficient to type:
python
and press Enter, and that’s it. In order to quit the terminal, use the
exit command, or press Control-d on an empty line.
Moving around. From my home, I can move to other directories using the
cd command. For instance, if I am in my home directory /home/stefano/
and I type:
cd Devel/sciprog/lectures
I will move to the directory:
/home/stefano/Devel/sciprog/lectures
The special path .. can be used to go back up one level, so if I am in:
/home/stefano/Devel/sciprog/lectures
and execute cd .., I end up in:
/home/stefano/Devel/sciprog
At any point in time, I can use the pwd command to print the path of the
directory I am currently in. For instance, from a newly opened terminal I
could do:
$ pwd
/home/stefano
$ cd Devel/sciprog/lectures
$ pwd
/home/stefano/Devel/sciprog/lectures
$ cd ..
/home/stefano/Devel/sciprog
If I use cd alone (not followed by a path), I will move to the home
directory, irregardless of where I currently am:
$ pwd
/home/stefano/Devel/sciprog/lectures
$ cd
/home/stefano
More information. Of course there are many more things you can do with/from the terminal (most of them, actually), but I won’t write about them. Feel free to consult an online tutorial, like this for further inputs.
Some useful commands:
| Command | Meaning |
|---|---|
pwd |
Print the directory I am currently in |
cd <path> |
Move to a different directory |
ls (<path>) |
List contents of the given directory |
mv <path> <path> |
Move a file/directory somewhere else |
cp <path> <path> |
Copie a file/directory |
rm <path> |
Delete a file/directory |
mkdir <path> |
Create a new directory somewhere |
cat <path> |
Print the contents of a file |
And some useful key presses:
| Keypress | Meaning |
|---|---|
Control-c |
Kill any running program (may not always work) |
Control-d |
Means “end-of-data”, useful for exiting the terminal. |
Control-l |
Cleans the terminal window. |
Tab |
Auto-completes the current command/path. |
↑, ↓ |
Recalls previously used commands. |
history |
Prints previously used commands on screen. |
Python: Basics¶
I will be teaching the Python language, version 2.7 (more on that later).
The biggest take-away message from this lecture is:
Danger
In order to learn Python, you have to write Python code.
The converse is also true, so:
Danger
If you do not write Python code, you will not learn Python.
Please keep this in mind.
The Interpreter¶
The term Python refers to the Python language, while python refers to
the Python interpreter.
In order to access the interpreter, type:
python
in a terminal window. You will get a prompt that looks like this:
Python 2.7.12+ (default, Sep 1 2016, 20:27:38)
[GCC 6.2.0 20160822] on linux2
Type "help", "copyright", "credits" or "license" for more information.
>>>
Now you are free to type your Python code into the interpreter, and it will be executed immediately. Try it out with:
print "hello, world!"
To quit the interpreter, either type:
quit()
or press Control-d on a blank line.
Warning
There are two versions of the Python language out there:
Python 2: this is the version we will be using. The current version of the Python 2 interpreter is
2.7.12.When we talk about Python, we refer to Python 2.
Python 3: this is a new and improved version of the language.
It is mostly compatible with Python 2: a lot of valid Python 2 code is also valid Python 3 code. Alas, there are also slight differences. I will not explain what these are.
The current version of the Python 3 interpreter is
3.5.2.
We will stick to Python 2 in this course.
Warning
There are some minor differences between the languages allowed by the different versions of the Python interpreter.
For instance, list comprehensions have been available since Python 2,
interpreter version 2.0, but generators (which work similarly) have
only been introduced in version 2.4.
Modules¶
A module is a text file hosting a bunch of Python code.
If you want to execute Python code written in a text file, you have to:
Make sure that the file has a
.pyextension, for instanceexample.py.Type:
python example.py
Warning
The .py extension is mandatory! If you use any other extension, the
interpreter will not recognize your text file as containing valid Python
code.
Let’s say that you wrote a module util.py, which includes a function
do_stuff(). In order to use that function from a different module, you
have to import the module, as follows:
import util
util.do_stuff()
The do_stuff() function is prefixed with the name of the module. This
way there is no name clash between functions (or other symbols) in different
modules.
Note
The name of the module (as seen from inside Python) is util, not
util.py!
Objects¶
An object represents a datum (for instance, a number, some text, a collection of things, etc.) as well as functions for operating on that datum.
Every object has:
- a type, which specifies what the kind of data represented by the object.
- a value, that is, the data itself.
In a way, the type is a form of meta-data (information about the data).
Python provides some elementary types:
| Type | Meaning | Domain | Mutable? |
|---|---|---|---|
bool |
Condition | True, False |
No |
int |
Integer | \(\{-2^{-63},\ldots,2^{63}-1\}\) | No |
long |
Integer | \(\mathbb{N}\) | No |
float |
Rational | \(\mathbb{Q}\) (more or less) | No |
str |
Text | Text | No |
list |
Sequence | Collections of things | Yes |
tuple |
Sequence | Collections of things | No |
dict |
Map | Maps between things | Yes |
Most of these should be pretty obvious. Later on, we will see how the more
complex ones (list, tuple and dict) mean.
Variables¶
Variables are references to objects. You can view them as names for the objects that they refer to.
An object is assigned to a variable with the assignment operator =:
pi = 3.1415926536
Here the variable pi refers to the object 3.1415926536. Informally,
we just decided to use the name pi to refer to the object 3.1415926536.
Warning
The name of the variable is arbitrary!
You can pick any name you like, as long as you follow some simple rules:
- the only allowed characters are upper-case digits (A-Z), lower-case
digits (a-z), numerical digits (0-9), and the underscore
_. - the name can not begin with a number.
The type of a variable is given by the type of the object it refers to. In the
previous example, the type of 3.1415926536 is float; as a consequence,
the type of pi is also float.
Warning
The variable does not contain the object; it merely contains a reference to the object.
To print the value of a variable (or rather, of its associated object), use
the print function:
var = "I am an example"
print var
To print the type of a variable (object), use the type function:
var = "I am an example"
print type(var)
Example. Just to wrap up, let’s create a new variable var:
var = 123
The name of var is var, its value is 123, and its type is int:
print var # 123
print type(var) # int
Example. A variable can be assigned multiple times:
var = 1
print var # 1
print type(var) # int
var = "MANLFKLGAENIFLGRKAATKEEAIRF"
print var # "MANLFKLGAENIFLGRKAATKEEAIRF"
print type(var) # str
var = 3.1415926536
print var # 3.1415926536
print type(var) # float
Note how the value and type of var changes after each assignment!
Example. Assignment also works between variables:
a = "I am an example"
print a
b = a
print b
Here the variable b is assigned the same object that is referenced by
a, that is, "I am an example".
Functions and Methods¶
Python provides a variety of functions and methods, for instance print()
(to print the value of an expression) and help() (to show the manual of
a given type, function, method, class, or module).
A function takes zero or more objects as inputs (its arguments), performs some operation on them, and returns an object (its result).
To invoke a function
f(), write:result = f(arg1, .., argN)
Here the variable
resultis assigned to the result of the function.A method is exactly like a function, except that it is provided by a given type. For instance, strings provide a
split()method, which can only be used in conjunction with strings; lists provide ajoin()method, which only works with lists. The other basic types (except for numbers) also provide their own methods.To invoke a method
t.m(), write:result = obj_or_var_for_type_t.m(arg1, ..., argN)
Warning
Python functions and mathematical functions share some similarities: informally, both evaluate their arguments (inputs) to produce a result (output).
There are some important differences though:
- Not all Python arguments require arguments. They may take none at all.
- Python functions may not return any result. (In which case they always
evaluate to
None). - Mathematical functions can only operate on their arguments, while Python functions can access additional resources.
- Mathematical functions can only modify the “external environment” through their result. Python functions can have side-effects.
For now, you can just use functions and methods already provided by Python; later on in the course we will write our own functions!
Documentation¶
In order to open the interactive manual of a type/object/variable, you can use
the help() function. For instance:
help(1)
shows the manual of the int type; you can achieve the same result with:
help(int)
and:
x = 1
help(x)
To open the manual of a given function or method, you can use the same procedure, for instance:
# Opens the manual of the len() function
help(len)
# Opens the manual of the split() method of the str type
help(str.split)
# help("some text".split)
To quit the manual, press q.
Python: Numbers¶
There are four distinct numeric types:
int: integers. (Limited precision.)long: long integers. (Unlimited precision.) They behave exactly like plainint‘s, but they can assume arbitrary values.float: floating point numbers. Also called floats, they represent real numbers. They are written with a decimal point dividing the integer and fractional parts. Floats may also be in scientific notation, with E or e indicating the power of 10.bool: Boolean conditions. They can assume only two values:TrueandFalse. They are used to represent the result of comparisons.
Example. Let’s create four variables, one for each type, and print them on the screen with print:
x = 10
y = 10L
z = 3.14
c = False
# Print the four variables
print x, y, z, c
# The same, adding some text
print "x =", x, "y =", y, "z =", z, "; the condition is", c
This syntax of print is valid for all variable types, not only numeric types.
Operations¶
All numeric types support default arithmetic operations:
- sum
+, difference-, product* - division
/, integer division//, remainder%. - power
**.
The type of the result of n operation m is the most “complex” type between the type of
n and m. The complexity hierarchy is:
bool < int < long < float
Example. Two examples of automatic conversion:
result = 1 * 1L # int * long
print result
print type(result) # long
result = 1.0 + 1L # float * long
print result
print type(result) # float
Warning
To avoid errors, you should choose the most appropriate type for your variables, so that the type of the result is sufficiently “complex” to represent the value of the result!
See for example the following GC-content exercise.
All numeric types support comparison operations:
- equal
==not equal!=(or<>) - strictly less than
<strictly greater than> - less than or equal``<=`` greater than or equal
>=
The expression has value True if the condition is satisfied, otherwise False.
The result of a comparison is always bool.
Example. Let’s consider:
na, nc, ng, nt = 2, 6, 50, 4
result = (na + nt) > (nc + ng)
print result
print type(result)
We can build complex conditions using Boolean operations:
a and bisTrueif bothaandbareTrue.a or bisTrueif at least one betweenaandbisTrue.not aisTrueonly ifaisFalse.
Warning
In this context, both a and b should be bool, otherwise the result may seem apparently bizarre.
Example. x > 12 and x < 34 both result bool, so we can combine them to obtain:
# int int
# | |
print (x > 12) and (x < 34)
# \______/ \______/
# bool bool
# \___________________/
# bool
or:
# int int
# | |
print (not (x > 12)) or (x < 34)
# \______/
# bool
# \____________/ \______/
# bool bool
# \________________________/
# bool
Examples¶
Example. Let’s calculate the zeros of the quadratic equation \(x^2 - 1 = 0\):
a, b, c = 1.0, 0.0, -1.0
delta = b**2 - 4*a*c
zero1 = (-b + delta**0.5) / (2 * a)
zero2 = (-b - delta**0.5) / (2 * a)
print zero1, zero2
Here we use x**0.5 to calculate the square root: \(\sqrt{x} = x^\frac{1}{2}\).
Example. We want to calculate the GC-content of a genomic region. We know that the region:
- Is 1521 nucleotides long.
- Contains 316 cytosines.
- Contains 235 guanines.
The GC-content is defined as \((g + c) / n\). To calculate the result, we may write:
n, c, g = 1521, 316, 235
gc_content = (c + g) / n
print gc_content
unfortunately, the result of this code is 0! Why?
The problem is that the resulting proportion, amounting to approximately 0.362, cannot be expressed by an integer: we need a rational number. Since both n and m
are integers, also gc_content = n / m will be an integer:
print type(gc_content)
As a consequence, 0.362 is approximated to the nearest integer: 0.
Oops!
To solve this problem, gc_content needs to be a float. We can do it in two ways:
Modifying the type of
n,morg:n, c, g = 1521.0, 316.0, 235.0 gc_content = (c + g) / n print gc_content
Using an explicit conversion to a
float:n, c, g = 1521, 316, 235 gc_content = float(c + g) / float(n) print gc_content
Example. To check that x falls in the interval A \(= [10,50]\)
let’s write:
x = 17
min_a, max_a = 10, 50
inside_a = (min_a <= x <= max_a)
print inside_a
or:
inside_a = ((x >= min_a) and (x <= max_a))
Assuming that inside_a, inside_b and inside_c indicate that x is contained
in interval A, B or C, respectively, we can create more complex conditions:
# x is in at least one interval
inside_at_least_one = inside_a or inside_b or inside_c
# x is in both A and B, but not in C
inside_a_and_b_but_not_c = inside_a and inside_b and (not inside_c)
Exercises¶
Create the following variables, checking that value and type are correct (using
printandtype):aandbwith values12and23as integers.canddwith values34and45as long integers.xandywith values21and14as floats.
Using
print(once), print:- All the above variables in the same line.
- All the above variables, separated by
;, on the same line. - The text “the product of
aandbisa * b”, replacinga,banda * bwith the variable values.
Find the value and the type of:
- The product of
aandb. - The difference between
candd. - The division of
xbyy. - The integer division of
abyb. - The integer division of
cbyd. - The integer division of
xbyy. - The product of
aandc. - The product of
bandy. - The product of
xandd. 2to the power0.2to the power100.2to the power1.2.2to the power-2.- The square root of
4. - The square root of
2.
- The product of
What is the difference between:
10 / 1210 / 12.010 // 1210 // 12.0
What is the difference between:
10 % 310 % 3.0
Using
pi = 3.141592and givenr = 2.5, calculate:- The circumference of a circle with radius
r: \(2 \pi r\). - The area of a circle with radius
r: \(\pi r^2\). - The volume of a sphere with radius
r: \(\frac{4}{3} \pi r^3\).
- The circumference of a circle with radius
Create 2 variables
a = 100andb = True. Using an adequate number of auxiliary variables (with arbitrary names!), give the value ofatoband viceversa.(Writing
a = Trueandb = 100is not sufficient!)Can it be done with only one auxiliary variable?
On the same strand of DNA there are 2 genes. The first includes i nucelotides from position 10 to position 20, the second nucleotides from position 30 to position 40. Let’s write this:
gene1_start, gene1_end = 10, 20 gene2_start, gene2_end = 30, 40
Given a variable
posrepresenting an arbitrary position on the DNA strand, write some comparisons to verify if:posis in the first gene.posis in the second gene.posis between the start of the first gene and the end of the second.posis between the start of the first gene and the end of the second, but not in any of the genes.posis before the start of the first gene or after the end of the second.posis inside one of the genes.posis distant no more than 10 nucleotides from the beginning of the first gene.
Given three Boolean variables
t,u, andv, write expressions that areTrueif and only if:t,u,vare all true.tis true oruis true, but not both.- Exactly one of the three variables is false.
- Exactly one of the three variables is true.
Exercise 1.4 from [book2]: Exponential Growth
Given optimal growth conditions, a single E. coli bacterium can divide within 20 minutes. If the conditions stay optimal, how many bacteria are there after 8 hours?
Python: Numbers (Solutions)¶
Solutions:
a = 12 b = 23 print a, b print type(a), type(b) # int, int c = 34L d = 45L print c, d print type(c), type(d) # long, long x = 21.0 y = 14.f print x, y print type(x), type(y) # float, float
Solutions:
print a, b, c, d, x, y print a, ";", b, ";", c, ";", ...
Solutions:
# simple operations: product = a * b # int * int print product print type(product) # int difference = c - d # long - log print difference print type(difference) # long # division and integer division between various numeric types: quotient = x / y # float / float print quotient print type(quotient) # float result = a // b # int // int print result print type(result) # int result = c // d # long // long print result print type(result) # long result = x // y # float // float print result print type(result) # float # more complex examples, where the result type # is the more complex type between the types # of the operands: result = a * c # int * long print result print type(result) # long result = b * y # int * float print result print type(result) # float result = x * d # float * long print result print type(result) # float # here the type is automatically defined # based on the magnitude of the result: result = 2**0 # int**int print result print type(result) # int result = 2**0 # int*int print result print type(result) # *** long!!! *** result = 2**1.2 # int*float print result print type(result) # float result = 2**-2 # int*int print result print type(result) # *** float!!! *** result = 4**0.5 # int*float print result print type(result) # float result = 2**0.5 # int*float print result print type(result) # float
Solutions:
>>> print 10 / 12 0 >>> print 10 / 12.0 0.833333333333 >>> print 10 // 12 0 >>> print 10 // 12.0 0.0
As you see, integer division is consistent with types: when it is applied to floats, the result is also a float, but truncated to the integer
0.Solutions:
>>> 10 % 3 1 >>> 10 % 3.0 1.0
As you see,
%returns the remainder of10 / 3:10 = 3*3 + 1 # ^ # remainder
Operand types don’t influence the value of the result, only its type.
Solution:
pi = 3.141592 r = 2.5 circumference = 2 * pi * r print circumference area = 2 * pi * r**2 print area area = 2 * pi * r * r print area volume = (4.0 / 3.0) * pi * r**3 print volume
Be careful:
4 / 3 != 4.0 / 3.0.Solution:
a, b = 100, True a2 = a b2 = b b = a2 a = b2 print a, b
or:
a, b = 100, True x = a a = b b = x print a, b
Solution:
gene1_start, gene1_end = 10, 20 gene2_start, gene2_end = 30, 40 # handy pictogram: # # 5' 3' # ~~~~~xxxxxxxx~~~~~xxxxxxx~~~~~> # 10 20 30 40 # \______/ \_____/ # gene_1 gene_2 # two options condition_1 = (10 <= pos <= 20) condition_1 = (pos >= 10 and pos <= 20) condition_2 = (30 <= pos <= 40) condition_3 = (10 <= pos <= 40) # two options condition_4 = condition_3 and not (condition_1 or condition_2) condition_4 = (20 <= pos <= 40) condition_5 = pos < 10 or pos > 40 # Be careful: # # pos < 10 and pos > 40 # # doesn't make sense: it's always False! condition_6 = condition_1 or condition_2 condition_7 = (0 <= pos <= 20)
Test the code with multiple values of
position, in order to check that conditions have been correctly formulated:Truewhen the position satisfies the condition, andFalseotherwise.Solution:
all_the_three = t and u and v t_or_u_but_not_both = (t or u) and not (t and u) # NOTE: here backslash at the end of lines are used to start a new line, you can ignore them. one_of_three_false = \ (t and u and not v) or \ (t and not u and v) or \ (not t and u and v) one_of_three_true = \ (t and not u and not v) or \ (not t and u and not v) or \ (not t and not u and v)
Again, the code needs to be tested with different values of
t,uandv. There are 8 possible combinations:t, u, v = False, False, False t, u, v = False, False, True t, u, v = False, True, False t, u, v = False, True, True # ...
Solution:
replication_time = 20 total_time= 480 final_bacteria = 2**(total_time//replication_time) print final_bacteria
Python: Strings¶
Strings are immutable objects representing text.
To define a string, we can write:
var = "text"
or, equivalently:
var = 'text'
To insert special characters, we need to perform escaping with a backslash
\:
path = "data\\fasta"
or use the prefix r (raw):
path = r"data\fasta"
Note
Here is a reference list of escape characters. You will probably only need
the most obvious ones, like \\, \n and \t.
| Escape Character | Meaning |
|---|---|
\\ |
Backslash |
\' |
Single-quote |
\" |
Double-quote |
\a |
ASCII bell |
\b |
ASCII backspace |
\f |
ASCII formfeed |
\n |
ASCII linefeed (also known as newline) |
\r |
Carriage Return |
\t |
Horizontal Tab |
\v |
ASCII vertical tab |
\N{name} |
Unicode character name (Unicode only!) |
\uxxxx |
Unicode 16-bit hex value xxxx (u’’ string only) |
\Uxxxxxxxx |
Unicode 32-bit hex value xxxxxxxx (u’’ string only) |
\ooo |
Character with octal value ooo |
\xhh |
Character with hex value hh |
To create a multi-line string, we can manually place the
newline character \n at each line:
sad_joke = "Time flies like an arrow.\nFruit flies like a banana."
print sad_joke
or we can use triple quotes:
sad_joke = """Time flies like an arrow.
Fruit flies like a banana."""
print sad_joke
Warning
print interprets special characters, while terminal echo doesn’t.
Try to write:
print path
and (from the interpreter):
path
In the rirst case, we see one slash (the escaping slash is automatically interpreted by print), in the second case we see two slashes (the escape slash is not interpreted).
The same if we print sad_joke.
String-Number conversion¶
We can convert a number into a string with str():
n = 10
s = str(n)
print n, type(n)
print s, type(s)
int() or float() perform the opposite conversion:
n = int("123")
q = float("1.23")
print n, type(n)
print q, type(q)
If the string doesn’t contain the correct numeric type, Python will give an error message:
int("3.14") # Not an int
float("ribosome") # Not a number
int("1 2 3") # Not a number
int("fifteen") # Not a number
Operations¶
| Result | Operator | Meaning |
|---|---|---|
bool |
== |
Check whether two strings are identical. |
int |
len(str) |
Return the length of the string |
str |
str + str |
Concatenate two strings |
str |
str * int |
Replicate the string |
bool |
str in str |
Check if a string is present in another string |
str |
str[int:int] |
Extract a sub-string |
Example. Let’s concatenate two strings:
string = "one" + " " + "string"
length = len(string)
print "the string:", string, "is", length, "characters long"
Another example:
string = "Python is hell!" * 1000
print "the string is", len(string), "characters long"
Warning
We cannot concatenate strings with other types. For example:
var = 123
print "the value of var is" + var
gives an error message. Two working alternatives:
print "the value of var is" + str(123)
or:
print "the value of var is", var
(In the second case we miss a space between is and 123.)
Example. The operator substring in string checks if
substring appears once or more times in string, for example:
string = "A beautiful journey"
print "A" in string # True
print "beautiful" in string # True
print "BEAUTIFUL" in string # False
print "ul jour" in string # True
print "Gengis Khan" in string # False
print " " in string # True
print " " in string # False
The result is always True or False.
Example. To extract a substring we can use indexes:
# 0 -1
# |1 -2|
# ||2 -3||
# ||| ... |||
alphabet = "abcdefghijklmnopqrstuvwxyz"
print alphabet[0] # "a"
print alphabet[1] # "b"
print alphabet[len(alphabet)-1] # "z"
print alphabet[len(alphabet)] # Error
print alphabet[10000] # Error
print alphabet[-1] # "z"
print alphabet[-2] # "y"
print alphabet[0:1] # "a"
print alphabet[0:2] # "ab"
print alphabet[0:5] # "abcde"
print alphabet[:5] # "abcde"
print alphabet[-5:-1] # "vwxy"
print alphabet[-5:] # "vwxyz"
print alphabet[10:-10] # "klmnop"
Warning
Extraction is inclusive with respect to the first index, but exclusive with respect to the second. In other words alphabet[i:j] corresponds to:
alphabet[i] + alphabet[i+1] + ... + alphabet[j-1]
Note that alphabet[j] is excluded.
Warning
Extraction return a new string, leaving the original unvaried:
alphabet = "abcdefghijklmnopqrstuvwxyz"
substring = alphabet[2:-2]
print substring
print alphabet # Is unvaried
Methods¶
| Result | Method | Meaning |
|---|---|---|
str |
str.upper() |
Return the string in upper case |
str |
str.lower() |
Return the string in lower case |
str |
str.strip(str) |
Remove strings from the sides |
str |
str.lstrip(str) |
Remove strings from the left |
str |
str.rstrip(str) |
Remove strings from the right |
bool |
str.startswith(str) |
Check if the string starts with another |
bool |
str.endswith(str) |
Check if the string ends with another |
int |
str.find(str) |
Return the position of a substring |
int |
str.count(str) |
Count the number of occurrences of a substring |
str |
str.replace(str, str) |
Replace substrings |
Warning
Methods return a new string, leaving the original unvaried (as with extraction):
alphabet = "abcdefghijklmnopqrstuvwxyz"
alphabet_upper = alphabet.upper()
print alphabet_upper
print alphabet # Is unvaried
Example. upper() and lower() are very simple:
text = "No Yelling"
result = text.upper()
print result
result = result.lower()
print result
Example. strip() variants are also simple:
text = " one example "
print text.strip() # equivalent to text.strip(" ")
print text.lstrip() # idem
print text.rstrip() # idem
print text # text is unvaried
Note that the space between "one" and "example" is never removed. We can specify more than one character to be removed:
"AAAA one example BBBB".strip("AB")
Example. The same is valid with startswith() and endswith():
text = "123456789"
print text.startswith("1") # True
print text.startswith("a") # False
print text.endswith("56789") # True
print text.endswith("5ABC9") # False
Example. find() returns the position of the first occurrence of a substring, or -1 if the substring never occurs:
text = "123456789"
print text.find("1") # 0
print text.find("56789") # 4
print text.find("Q") # -1
Example. replace() returns a copy of the string where a substring is replaced with another:
text = "if roses were rotten, then"
print text.replace("ro", "gro")
Example. Given this unformatted string of aminoacids:
sequence = ">MAnlFKLgaENIFLGrKW "
To increase uniformity, we want to remove the ">" character, remove spaces and finally convert everything to upper case:
s1 = sequence.lstrip(">")
s2 = s2.rstrip(" ")
s3 = s2.upper()
print s3
Alternatively, all in one step:
print sequence.lstrip(">").rstrip(" ").upper()
Why does it work? Let’s write it with brackets:
print ( ( sequence.lstrip(">") ).rstrip(" ") ).upper()
\_____________________/
str
\_____________________________________/
str
\_____________________________________________/
str
As you can see, the result of each method is a string (as s1, s2 e s3 in the example above);
and we can invoke string methods.
Exercises¶
How can I:
Create a string consisting of five spaces only.
Check whether a string contains at least one space.
Check whether a string contains exactly five (arbitrary) characters.
Create an empty string, and check whether it is really empty.
Create a string that contains one hundred copies of
"Python is great".Given the strings
"but cell","biology"and"is way better", compose them into the string"but cell biology is way better".Check whether the string
"12345"begins with1(the character, not the number!)Create a string consisting of a single character
\. (Check whether the output matches using both the echo of the interpreter andprint, and possibly also withlen())Check whether the string
"\\"contains one or two backslashes.Check whether a string (of choice) begins or ends by
\.Check whether a string (of choice) contains
xat least three times at the beginning and/or at the end. For instance, the following strings satisfy the desideratum:"x....xx" # 1 + 2 >= 3 "xx....x" # 2 + 1 >= 3 "xxxx..." # 4 + 0 >= 3
while these do not:
"x.....x" # 1 + 1 < 3 "...x..." # 0 + 0 < 3 "......." # 0 + 0 < 3
Given the string:
s = "0123456789"
which of the following extractions are correct?
s[9]s[10]s[:10]s[1000]s[0]s[-1]s[1:5]s[-1:-5]s[-5:-1]s[-1000]
Create a two-line string that contains the two following lines of text literally, including all the special characters and the implicit newline character:
never say “never”!
said the sad turtle
Given the strings:
string = "a 1 b 2" digit = "DIGIT" character = "CHARACTER"
replace all the digits in the variable
stringwith the text provided by the variabledigit, and all alphabetic characters with the content of the variablecharacter.The result should look like this:
"CHARACTER DIGIT CHARACTER DIGIT"You are free to use auxiliary variables to hold any intermediate results, but do not need to.
Given the following multi-line sequence:
chain_a = """SSSVPSQKTYQGSYGFRLGFLHSGTAKSVTCTYSPALNKM FCQLAKTCPVQLWVDSTPPPGTRVRAMAIYKQSQHMTEVV RRCPHHERCSDSDGLAPPQHLIRVEGNLRVEYLDDRNTFR HSVVVPYEPPEVGSDCTTIHYNYMCNSSCMGGMNRRPILT IITLEDSSGNLLGRNSFEVRVCACPGRDRRTEEENLRKKG EPHHELPPGSTKRALPNNT"""
which represents the aminoacid sequence of the DNA-binding domain of the Tumor Suppressor Protein TP53 , answer the following questions.
- How many lines does it hold?
- How long is the sequence? (Do not forget to ignore the special characters!)
- Remove all newline characters, and put the result in a new variable
sequence. - How many cysteines
"C"are there in the sequence? How many histidines"H"? - Does the chain contain the sub-sequence
"NLRVEYLDDRN"? In what position? - How can I use
find()and the sub-string extraction[i:j]operators to extract the first line fromchain_a?
Given (a small portion of) the tertiary structure of chain A of the TP53 protein:
structure_chain_a = """SER A 96 77.253 20.522 75.007 VAL A 97 76.066 22.304 71.921 PRO A 98 77.731 23.371 68.681 SER A 99 80.136 26.246 68.973 GLN A 100 79.039 29.534 67.364 LYS A 101 81.787 32.022 68.157"""
Each line represents an \(C_\alpha\) atom of the backbone of the structure. Of each atom, we know: - the aminoacid code of the residue - the chain (which is always
"A"in this example) - the position of the residue within the chain (starting from the N-terminal) - and the \(x, y, z\) coordinates of the atomExtract the second line using
find()and the extraction operator. Put the line in a new variableline.Extract the coordinates of the second residue, and put them into three variables
x,y, andz.Extract the coordinates from third residue as well, putting them in different variables
x_prime,y_prime,z_primeCompute the Euclidean distance between the two residues:
\(d((x,y,z),(x',y',z')) = \sqrt{(x-x')^2 + (y-y')^2 + (z-z')^2}\)
Hint: make sure to use
floatnumbers when computing the distance.
Given the following DNA sequence, part of the BRCA2 human gene:
dna_seq = """GGGCTTGTGGCGCGAGCTTCTGAAACTAGGCGGCAGAGGCGGAGCCGCT GTGGCACTGCTGCGCCTCTGCTGCGCCTCGGGTGTCTTTT GCGGCGGTGGGTCGCCGCCGGGAGAAGCGTGAGGGGACAG ATTTGTGACCGGCGCGGTTTTTGTCAGCTTACTCCGGCCA AAAAAGAACTGCACCTCTGGAGCGG"""
- Calculate the GC-content of the sequence
- Convert the DNA sequence into an RNA sequence
- Assuming that this sequence contains an intron ranging from nucleotide 51 to nucleotide 156, store the sequence of the intron in a string, and the sequence of the spliced transcript in another string.
Python: Strings (Solutions)¶
Note
Later on in the solutions, I will sometimes use the backslash character
\ at the end of a line.
When used this way, \ tells Python that the command continues on the
following line, allowing to break long commands over multiple lines.
Solutions:
Solution:
# 12345 text = " " print text print len(text)
Solution:
at_least_one_space = " " in text # check whether it works print " " in "nospaceatallhere" print " " in "onlyonespacehere--> <--" print " " in "more spaces in here"
Solution:
exactly_5_characters = len(text) == 5 # check whether it works print len("1234") == 5 print len("12345") == 5 print len("123456") == 5
Solution:
empty_string = "" print len(empty_string) == 0
Solution:
base = "Python is great" repeats = base * 100 # check whether the length is correct print len(repeats) == len(base) * 100
Solution:
part_1 = "but cell" part_2 = "biology" part_3 = "is way better" text = (part_1 + part_2 + part_3) * 1000
Let’s try this:
start_with_1 = "12345".startswith(1)
but Python gives an error message:
Traceback (most recent call last): File "<stdin>", line 1, in <module> TypeError: startswith first arg must be str, unicode, or tuple, not int # ^^^^^^^^^^^^^^^^^^^^^ ^^^^^^^
The error message, see highlighted parts, says that
startswith()requires the argument to be a string, non an int as in our case:1, is an int.The solution is:
start_with_1 = "12345".startswith("1") print start_with_1
the value is
True, as expected.Solution:
string = "\\" string print string print len(string) # 1
alternatively:
string = r"\" string print string print len(string) # 1
Already checked before, the answer is no. Anyway:
backslash = r"\" print backslash*2 in "\\" # False
First method:
backslash = r"\" condition = text.startswith(backslash) or \ text.endswith(backslash)
Second method:
condition = (text[0] == backslash) or \ (text[-1] == backslash)
Solution:
condition = \ text.startswith("xxx") or \ (text.startswith("xx") and text.endswith("x")) or \ (text.startswith("x") and text.endswith("xx")) or \ text.endswith("xxx")
It’s worth to check the condition using the examples provided in the exercise.
Solution:
s = "0123456789" print len(s) # 10
Which of the following extractions are correct?
s[9]: correct, extracts the last character.s[10]: invalid.s[:10]: corrett, extracts all characters (remember that the second index,10in this case, is exclusive.)s[1000]: invalid.s[0]: correct, extracts the first character.s[-1]: correct, extracts the last character.s[1:5]: correct, ectracts from the 2nd to the 6th character.s[-1:-5]: corrects[-5:-1]: correct, but nothing is extracted (indexes are inverted!)s[-1000]: invalid.
Solution (one of two possible solutions):
text = """never say \"never!\" \said the sad turtle."""
Solution:
string = "a 1 b 2 c 3" digit = "DIGIT" character = "CHARACTER" result = string.replace("1", digit) result = result.replace("2", digit) result = result.replace("3", digit) result = result.replace("a", character) result = result.replace("b", character) result = result.replace("c", character) print result # "CHARACTER DIGIT CHARACTER ..."
In one line:
print string.replace("1", digit).replace("2", digit) ...
Solution:
chain_a = """SSSVPSQKTYQGSYGFRLGFLHSGTAKSVTCTYSPALNKM FCQLAKTCPVQLWVDSTPPPGTRVRAMAIYKQSQHMTEVV RRCPHHERCSDSDGLAPPQHLIRVEGNLRVEYLDDRNTFR HSVVVPYEPPEVGSDCTTIHYNYMCNSSCMGGMNRRPILT IITLEDSSGNLLGRNSFEVRVCACPGRDRRTEEENLRKKG EPHHELPPGSTKRALPNNT""" num_lines = chain_a.count("\n") + 1 print num_lines # 6 # NOTE: we want to know the length of the actual *sequence*, non the length of the *string* length_sequence = len(chain_a) - chain_a.count("\n") print length_sequenza # 219 sequence = chain_a.replace("\n", "") print len(chain_a) - len(sequence) # 5 (giusto) print len(sequence) # 219 num_cysteine = sequence.count("C") num_histidine = sequence.count("H") print num_cysteine, num_histidine # 10, 9 print "NLRVEYLDDRN" in sequence # True print sequence.find("NLRVEYLDDRN") # 106 # let's check print sequence[106 : 106 + len("NLRVEYLDDRN")] # "NLRVEYLDDRN" index_first_newline = chain_a.find("\n") first_line = chain_a[:index_first_newline] print first_line
Solution:
structure_chain_a = """SER A 96 77.253 20.522 75.007 VAL A 97 76.066 22.304 71.921 PRO A 98 77.731 23.371 68.681 SER A 99 80.136 26.246 68.973 GLN A 100 79.039 29.534 67.364 LYS A 101 81.787 32.022 68.157""" # I use a variable with a shorter name chain = structure_chain_a index_first_newline = chain.find("\n") index_second_newline = chain[index_first_newline + 1:].find("\n") index_third_newline = chain[index_second_newline + 1:].find("\n") print index_first_newline, index_second_newline, index_third_newline second_line = chain[index_first_newline + 1 : index_second_newline] print second_line # "VAL A 97 76.066 22.304 71.921" # | | | | | | # 01234567890123456789012345678 # 0 1 2 x = second_line[9:15] y = second_line[16:22] z = second_line[23:] print x, y, z # NOTE: they are all strings third_line = chain[index_second_newline + 1 : index_third_newline] print third_line # "PRO A 98 77.731 23.371 68.681" # | | | | | | # 01234567890123456789012345678 # 0 1 2 x_prime = third_line[9:15] y_prime = third_line[16:22] z_prime = third_line[23:] print x_prime, y_prime, z_prime # NOTE: they are all strings # we should convert all variables to floats, in order to calculate distances x, y, z = float(x), float(y), float(z) x_prime, y_prime, z_prime = float(x_prime), float(y_prime), float(z_prime) diff_x = x - x_prime diff_y = y - y_prime diff_z = z - z_prime distance = (diff_x**2 + diff_y**2 + diff_z**2)**0.5 print distance
The solution is way simpler using
split():lines = chain.split("\n") second_line = lines[1] third_line = lines[2] words = second_line.split() x, y, z = float(words[-3]), float(words[-2]), float(words[-1]) words = third_line.split() x_prime, y_prime, z_prime = float(words[-3]), float(words[-2]), float(words[-1]) distance = ((x - x_prime)**2 + (y - y_prime)**2 + (z - z_prime)**2)**0.5
Solutions:
Solution:
dna_seq = dna_seq.replace("\n", "") # Remove newline characters length = len(dna_seq) # Calculate length ng = dna_seq.count("G") # Calculate the number of Gs nc = dna_seq.count("C") # Calculate the number of Cs gc_cont = (ng + nc)/float(length) # Calculate the GC-content
Solution:
rna_seq = dna_seq.replace("T","U")
Solution:
intron = dna_seq[50:156] # Careful with indexes exon1 = dna_seq[:50] # Careful with indexes exon2 = dna_seq[156:] # Careful with indexes spliced = exon1+exon2
Python: Lists¶
Lists are ordered sequences of arbitrary elements (objects).
Lists are defined using square brackets, as follows:
# A list of integers (notice that the 1 appears twice)
integers = [1, 2, 3, 1]
# A list of strings
uniprot_proteins = ["Y08501", "Q95747"]
# A list of heterogeneous objects
things = ["Y08501", 0.13, "Q95747", 0.96]
# A list of lists
two_level_list = [
["Y08501", 120, 520],
["Q95747", 550, 920],
]
# An empty list
empty = []
# A list containing two empty lists
a_weird_list = [ [], [] ]
Operations¶
| Returns | Operator | Meaning |
|---|---|---|
bool |
== |
Check whether two lists are identical |
bool |
!= |
Check whether two lists are different |
int |
len(list) |
Compute the length of a list |
list |
list + list |
Concatenate two list (returns a new list) |
list |
list * int |
Replicate a list multiple times |
bool |
element in list |
Check whether an element appears in the list |
list |
list[int:int] |
Extracts a sub-list |
list |
list[int] = object |
Assigns a new value to an element |
list |
range(int, [int]) |
Compute the integers in a given range |
Lists offer almost the same operators as strings, with a couple of additions.
Example. range() returns a list of integers in a given range:
>>> numbers = range(5)
>>> print numbers
[0, 1, 2, 3, 4]
>>> numbers = range(0, 5)
>>> print numbers
[0, 1, 2, 3, 4]
>>> numbers = range(2, 4)
>>> print numbers
[2, 3]
>>> numbers = range(4, 2) # the range is backwards!
>>> print numbers
[]
Example. Just like with strings, you can extract an element or a range of elements from a list:
>>> numbers = range(10)
>>> first_element = numbers[0]
>>> print first_element
0
>>> last_element = numbers[-1]
>>> print last_element
9
>>> the_other_elements = numbers[1:-1]
>>> print the_other_elements
[1, 2, 3, 4, 5, 6, 7, 8]
Example. The assignment operator assigns a new value to an already existing list element. It is more or less the opposite of the extraction operator. A couple of examples:
>>> numbers = range(5)
>>> print numbers
[0, 1, 2, 3, 4]
>>> numbers[0] = "first"
>>> print numbers
["first", 1, 2, 3, 4]
>>> numbers[-1] = "last"
>>> print numbers
["first", 1, 2, 3, "last"]
>>> numbers[len(numbers)/2] = "middle"
>>> print numbers
["first", 1, "middle", 3, "last"]
If the index is out-of-bounds, the assignment raises an error:
>>> numbers = range(5)
>>> numbers[100] = "out-of-bounds"
Traceback (most recent call last):
File "<stdin>", line 1, in <module>
IndexError: list assignment index out of range
Warning
The assignment operator does not change the length of the list!
It modifies an existing element (at a given position); it does not add a new element.
Warning
Lists are ordered: the order of the elements matters:
[1, 2, 3] != [3, 2, 1]
Lists are not sets: objects may appear more than once:
[3, 3, "a", "a"] != [3, "a"]
Exercises¶
Create an empty list using the bracket notation. Check whether it is really empty using
len().Create a list with the first five non-negative integers using
range().Create a list with one hundred
0elements.Hint: note the replication operator.
Given:
list_1 = range(10) list_2 = range(10, 20)
concatenate the two lists, and assign the result to a new variable
full_list. Use the equality comparison operator==to check whether it matches the result ofrange(20).Create a list of three strings:
"I am","a","list". Then print the type and length of the three elements (manually, one by one).Given the list:
list = [0.0, "b", [3], [4, 5]]
- How long is it?
- What is the type of the first element of
list? - How long is the second element of
list? - How long is the third element of
list? - What is the value of the last element of
list? How long is it? - Does the list contain an element
"b"? - Does the list contain an element
4?
What is the difference between these “lists”?:
list_1 = [1, 2, 3] list_2 = ["1", "2", "3"] list_3 = "[1, 2, 3]"
Hint: is the third one actually a list?
Which of the following code fragments are wrong?
list = []list = [}list = [[]]list = [1 2 3]list = range(3),element = list[3]list = range(3),element = list[-1]list = range(3),sublist = list[0:2]list = range(3),sublist = list[0:3]list = range(3),sublist = list[0:-1]list = range(3),list[2] = "two"list = range(3),list[3] = "three"list = range(3),list[-1] = "three"list = range(3),list[1.2] = "one point two"list = range(3),list[1] = ["protein1", "protein2"]
Given the list:
matrix = [ [1, 2, 3], # <-- 1st row [4, 5, 6], # <-- 2nd row [7, 8, 9], # <-- 3rd riga ] # ^ ^ ^ # | | | # | | +-- 3rd column # | +----- 2nd column # +-------- 1st column
How can I:
- Extract the first row?
- Extract the second element of the first row?
- Sum all elements of the first row? (Perform the sum manually)
- Create a new list containing the elements of the second column? (Manually)
- Create a new list containing the elements of the diagonal? (Manually)
- Create a list by concatenating the first, second, and third rows?
Methods¶
| Returns | Method | Meaning |
|---|---|---|
None |
list.append(object) |
Add a new element at the end of the list |
None |
list.extend(list) |
Add several new elements at the end of the list |
None |
list.insert(int,object) |
Add a new element at some given position |
None |
list.remove(object) |
Remove the first occurrence of an element |
None |
list.reverse() |
Invert the order of the elements |
None |
list.sort() |
Sort the elements |
int |
list.count(object) |
Count the occurrences of an element |
Warning
All list methods (except count()):
- Modify the input list
- Do not have a return value (they return
None)
In other words, they behave the exact opposite of string methods!
As a consequence, if I do:
list = range(10)
print list
result = list.append(10)
print list
print result
the list is modified in the process and result will be None. The
same is true for all other methods (except count()).
This may look a bit surprising, and it is easy to mistakenly expect
append() or reverse() to return a list. They do not.
By the way, this is the reason why we can not write code like:
list = []
list.append(1).append(2).append(3)
because the first append() does not return a list (it returns
None, and None of course does not support the append() method
– so the second append() always fails: Python raises an
error.)
Example. append() adds a new element at the end of a list:
list = range(10)
print list # [0, 1, 2, ..., 9]
print len(list) # 10
list.append(10)
print list # [0, 1, 2, ..., 9, 10]
print len(list) # 11
list.append(11)
print list # [0, 1, 2, ..., 9, 10, 11]
print len(list) # 12
Note how list changes in the process.
Example. extend() adds several new elements at the end of a list:
list = range(10)
list.extend(range(10,20))
print list # [0, 1, 2, ..., 19]
print len(list) # 20
Example. insert() adds a new element at an arbitrary position:
list = range(10)
print list # [0, 1, ..., 9]
print len(list) # 10
list.insert(2, "marker")
print list # [0, 1, "marker", 3, ..., 9]
print len(list) # 11
Example. Contrary to append(), insert() and extend(), list
concatenation does not modify the original list. You get a new list
instead:
list_1 = range(0, 10)
list_2 = range(10, 20)
# let's use the concatenation operator
full_list = list_1 + list_2
print list_1, "+", list_2, "->", full_list
# now let's use extend() instead
full_list = list_1.extend(list_2)
print list_1
print list_2
print full_list
Note how with extend(), list_1 changes while full_list is None,
as expected.
Example. remove() removes the first occurrence of a given value:
list = ["a", "list", "not", "a", "string"]
# ^^^ ^^^
list.remove("a")
print list # ["list", "not", "a", "string"]
Example. sort() and reverse() reorder the elements in the list:
list = [3, 2, 1, 5, 4]
list.reverse()
print list # [4, 5, 1, 2, 3]
list.sort()
print list # [1, 2, 3, 4, 5]
It also works with strings (it defaults to lexicographic ordering). Check this out:
list = ["AC", "GT"]
print list
list.reverse()
print list # ["GT", "AC"]
list.sort()
print list # ["AC", "GT"]
Example. count() returns the number of occurrences of a value in
a list:
list = ["a", "c", "g", "t", "a"]
num_a = list.count("a")
num_g = list.count("g")
print num_a, num_g # 2, 1
Of course, count() does not modify the original list.
Warning
A technical note. Recall that lists are mutable, and that (like all variables) they contain references to objects, not the objects themselves.
So what? Here are a few examples showing what are the consequences of the previous observation.
Example. This code:
sublist = range(5)
list = [sublist]
print list
creates a new list list that “contains” another list sublist. When
I modify sublist (which is mutable), I end up modifying list:
sublist.append(5)
print sublist
print list
Example. Another interesting case:
list = range(5)
print list
not_a_copy = list
print not_a_copy
# here both list and not_a_copy end up referring to the same
# object, so if I change list, I also end up changing
# not_a_copy!
list.append(5)
print list
print not_a_copy
In order to create a real, independent copy of a list, I have to use the extraction operator (or list comprehension, as we will see), as follows:
list = range(5)
print list
real_copy = list[:]
# or: real_copy = [elem for elem in list]
print real_copy
list.append(5)
print list
print real_copy
Exercises¶
Create a new empty list
list. Then add an integer, a string, and another list.Starting from the list
list = range(3)(reset it after every bullet point!), what happens when I do:#. ``list.append(3)`` #. ``list.append([3])`` #. ``list.extend([3])`` #. ``list.extend(3)`` #. ``list.insert(0, 3)`` #. ``list.insert(3, 3)`` #. ``list.insert(3, [3])`` #. ``list.insert([3], 3)``
What is the difference between:
list = [] list.append(range(10)) list.append(range(10, 20))
and:
list = [] list.extend(range(10)) list.extend(range(10, 20))
How long is
listin the two cases?What does this code do?:
list = [0, 0, 0, 0] list.remove(0)
What does this code do?:
list = [1, 2, 3, 4, 5] list.reverse() list.sort()
Is it equivalent to the following code?:
list = [1, 2, 3, 4, 5] list.reverse().sort()
Given the list:
list = range(10)
create a new list
reversed_listwith the same elements aslist, but in reversed order usingreverse().listmust not be changed in the process.Given the list list:
motifs = [ "KSYK", "SVALVV" "GVTGI", "VGSSLAEVLKLPD", ]
create a new list
sorted_motifswith the same elements asmotifs, but sorted alphanumerically usingsort().motifsmust not be changed in the process.
String-List Methods¶
| Returns | method | Meaning |
|---|---|---|
list-of-str |
str.split(str) |
Split a string into a list of strings (words) |
str |
str.join(list-of-str) |
Joins a list of strings (words) into a string |
Example. We have a multi-line string taken from a PDB structure file:
structure_chain_a = """SER A 96 77.253 20.522 75.007
VAL A 97 76.066 22.304 71.921
PRO A 98 77.731 23.371 68.681
SER A 99 80.136 26.246 68.973
GLN A 100 79.039 29.534 67.364
LYS A 101 81.787 32.022 68.157"""
Let’s split the multi-line string into a list of single-line strings, for ease of processing:
lines = structure_chain_a.split("\n")
print lines[0]
print lines[1]
# ...
Now we can extract for, say, the second line, all its words:
words = lines[1].split()
print words
It is now pretty easy to extract the coordinates of the residue:
coords = words[-3:]
print coords
Example. join() is essentially the inverse operation:
list_of_strings = [
">1A3A:A|PDBID|CHAIN|SEQUENCE",
"MANLFKLGAENIFLGRKAATKEEAIRFA",
]
multiline_string = "\n".join(list_of_strings)
print multiline_string
It is not a super-interesting method, but it can be useful for printing pretty formatted text.
Warning
Note that join() takes a list of strings! This won’t work:
" ".join([1, 2, 3])
Exercises¶
Given the text:
text = """The Wellcome Trust Sanger Institute is a world leader in genome research."""
create a list of string that includes all of the words (i.e. substrings separated by spaces) of
text.Then print how many words there are.
The table below:
tabella = [ "protein | database | domain | start | end", "YNL275W | Pfam | PF00955 | 236 | 498", "YHR065C | SMART | SM00490 | 335 | 416", "YKL053C-A | Pfam | PF05254 | 5 | 72", "YOR349W | PANTHER | 353 | 414", ]
(taken from Saccharomyces Genome Database) represents a list of domains that have been identified in a given yeast protein.
Each row is a single domain instance (except the first).
Use
split()to obtain the list of titles of the various columns (from the first row), making sure that the column names contain no spurious space characters.Hint:
strip()is not necessary; it is sufficient to usesplit()correctly.Given the list of strings:
words = ["word_1", "word_2", "word_3"]
build, using
join()together with an appropriate delimiter, the following strings:"word_1 word_2 word_3""word_1,word_2,word_3""word_1 e word_2 e word_3""word_1word_2word3"r"word_1\word_2\word_3"
Given the list of strings:
random_sentences = [ "Taci. Su le soglie", "del bosco non odo", "parole che dici", "umane; ma odo", "parole piu' nuove", "che parlano gocciole e foglie", "lontane." ]
use
join()to create a new multi-line stringpoem. The expected result is:>>> print poem Taci. Su le soglie del bosco non odo parole che dici umane; ma odo parole piu' nuove che parlano gocciole e foglie lontane.
Hint: what delimiter should I use?
List Comprehension¶
The list comprehension operator allows to filter or transform a list.
Warning
The original list is left unchanged. A new list is created instead.
As a filter. Given an arbitrary list original, I can create a new
list that only contains those elements of original that satisfy a given
condition.
The abstract syntax is:
filtered = [element
for element in original
if condition(element)]
Here condition() is arbitrary. Let’s see a few examples.
Example. Let’s create a list with the even numbers in the range [0, 99]:
numbers = range(100
even_numbers = [n
for n in numbers
if n % 2 == 0]
print even_numbers
Example. Given a list of DNA sequences:
sequences = ["ACTGG", "CCTGT", "ATTTA", "TATAGC"]
we keep only those sequences that contain at least one adenosine:
sequences_with_a = [sequence
for sequence in sequences
if "A" in sequence]
print sequences_with_a
If we want only those with no adenosine, we can invert the condition:
sequences_without_a = [sequence
for sequence in sequences
if not "A" in sequence]
print sequences_without_a
Example. When no condition is given, no filtering is performed:
list = range(5)
print list
list_2 = [element for element in list]
print list_2
The above code creates a copy of the original list list:
****
Example. This list describes a gene regulation network:
triples = [
["G1C2W9", "G1C2Q7", 0.2],
["G1C2W9", "G1C2Q4", 0.9],
["Q6NMS1", "G1C2W9", 0.8],
# ^^^^^^ ^^^^^^ ^^^
# gene1 gene2 correlation
]
Each “triple” has three elements: two A. Thaliana genes, and a measure of correlation of their expression (given by, say, a microarray experiment).
I can use a list comprehension to keep only the pairs of genes with high correlation:
high_correlation_genes = \
[triple[:-1] for triple in triples
if triple[-1] > 0.75]
I can also keep only those genes that are highly correlated with the
"G1C2W9" gene:
threshold = 0.75
interesting_genes = \
[triple[0] for triple in triples
if triple[1] == "G1C2W9" and triple[-1] >= threshold] + \
[triple[1] for triple in triples
if triple[0] == "G1C2W9" and triple[-1] >= threshold]
Warning
The name of the “temporary” variable holding the current element (in
the examples above, n, sequence and triple, respectively)
is arbitrary.
This code:
list = range(10)
print [x for x in list if x > 5]
is perfectly identical to this code:
list = range(10)
print [y for y in list if y > 5]
The name of the variable, x or y, does not make any difference.
You are free to pick any name you like.
As a transformation. Given an arbitrary list original, I can use a
list comprehension to also transform the elements in the list in some way.
The abstract syntax is:
transformed = [transform(element)
for element in original]
The transformation transform() is arbitrary.
Example. Given the list:
numbers = range(10)
let’s create a new list with their doubles:
doubles = [n * 2 for n in numbers]
# ^^^^^
# transformation
print doubles
Example. Given a list of paths:
paths = ["aatable", "fasta.1", "fasta.2"]
let’s add a prefix "data/" to each and every element:
prefixed_paths = ["data/" + path for path in paths]
# ^^^^^^^
# transformation
print prefixed_paths
Example. Given the list of primary sequences:
sequences = [
"MVLTIYPDELVQIVSDKIASNK",
"GKITLNQLWDIS",
"KYFDLSDKKVKQFVLSCVILKKDIE",
"VYCDGAITTKNVTDIIGDANHSYS",
]
let’s compute the length of each sequence, and save them in another list:
lengths = [len(seq) for seq in sequences]
print lengths
Example. Given the list of strings:
atoms = [
"SER A 96 77.253 20.522 75.007",
"VAL A 97 76.066 22.304 71.921",
"PRO A 98 77.731 23.371 68.681",
"SER A 99 80.136 26.246 68.973",
"GLN A 100 79.039 29.534 67.364",
"LYS A 101 81.787 32.022 68.157",
]
which represents (part of) the 3D structure of a protein chain, I want to
compute a list of lists which should hold, for each residue (that is, for
every row of atoms), its coordinates:
coords = [row.split()[-3:] for row in atoms]
The result is:
>>> print coords
[
["77.253", "20.522", "75.007"],
["76.066", "22.304", "71.921"],
["77.731", "23.371", "68.681"],
["80.136", "26.246", "68.973"],
["79.039", "29.534", "67.364"],
["81.787", "32.022", "68.157"],
]
Jointly transforming and filtering.* Given a list original, I can
both transform and filter its elements jointly using the complete version
of the list comprehension operator.
The abstract syntax is:
new_list = [transform(element)
for element in original
if condition(element)]
Example. Given the integers from 0 to 99, I want to keep only the even ones and divide them by 3:
result = [n / 3.0
for n in range(100)
if n % 2 == 0]
print result
Example. Given the list of strings:
atoms = [
"SER A 96 77.253 20.522 75.007",
"VAL A 97 76.066 22.304 71.921",
"PRO A 98 77.731 23.371 68.681",
"SER A 99 80.136 26.246 68.973",
"GLN A 100 79.039 29.534 67.364",
"LYS A 101 81.787 32.022 68.157",
]
we used:
coords = [row.split()[-3:] for row in atoms]
to obtain:
>>> print coords
[
["77.253", "20.522", "75.007"],
["76.066", "22.304", "71.921"],
["77.731", "23.371", "68.681"],
["80.136", "26.246", "68.973"],
["79.039", "29.534", "67.364"],
["81.787", "32.022", "68.157"],
]
We now make things more complex: we only want the coordinates of the serines. Let’s write:
coords = [row.split()[-3:] for row in atoms
if row.split()[0] == "SER"]
Exercises¶
Given the list:
list = range(100)
Create a new list
list_plus_3that holds the elements oflistplus3. The expected result is:[3, 4, 5, ...]
Create a new list
oddsthat holds only the odd elements inlist. The expected result is:[1, 3, 5, ...]
Hint: adapt one of the previous examples.
Create a new list
oppositesthat holds the arithmetical opposites (the opposite of \(x\) is \(-x\)) of the elements inlist. The expected result is:[0, -1, -2, ...]
Create a new list
inversesthat holds the arithmetical inverse (the inverse of \(x\) is \(\frac{1}{x}\)) of the elements inlist.Make sure to skip those elements that have no inverse (like
0).The expected result is:
[1, 0.5, 0.33334, ...]
Hint: skip = filter out.
Create a new list with only the first and last element of
list. The expected result is:[0, 99]
Hint: is a list comprehension required?
Create a new list with all elements of
list, except the first and the last. The expected result is:[1, 2, ..., 97, 98]
Count how many odd numbers there are in
list. They should be50.Hint: use a list comprehension plus... something else.
Create a new list holding all elements of
list, divided by 5. The expected result is:[0.0, 0.2, 0.4, ...]
Hint: make sure that the results are
float!Create a new list holding only the multiples of
5appearing inlist; the multiples should be divided by5. The expected result is:[0.0, 1.0, 2.0, ..., 19.0]
Create a new list
list_of_stringscontaining the same elements aslist, but converted into strings. The expected result is:["0", "1", "2", ...]
Count how many strings in
list_of_stringsrepresent an odd number. The expected result is, again,50.Create a string that contains all the elements in
list, separated by spaces. The expected result is:"0 1 2 ..."Hint: use a list comprehension plus... something else.
For each of the following bullet points, write two list comprehensions to convert from
list_1tolist_2and vice versa.list_1 = [1, 2, 3] list_2 = ["1", "2", "3"]
list_1 = ["name", "surname", "age"] list_2 = [["name"], ["surname"], ["age"]]
list_1 = ["ACTC", "TTTGGG", "CT"] list_2 = [["actc", 4], ["tttgggcc", 6], ["ct", 2]]
Given the list:
list = range(10)
which of the following code fragments are correct? What do they compute?
[x for x in list][y for y in list][y for x in list]["x" for x in list][str(x) for x in list][x for str(x) in list][x + 1 for x in list][x + 1 for x in list if x == 2]
Let’s consider the string:
clusters = """\ >Cluster 0 0 >YLR106C at 100.00% >Cluster 50 0 >YPL082C at 100.00% >Cluster 54 0 >YHL009W-A at 90.80% 1 >YHL009W-B at 100.00% 2 >YJL113W at 98.77% 3 >YJL114W at 97.35% >Cluster 52 0 >YBR208C at 100.00% """
extracted from the output of a clustering algorithm (CD-HIT) applied to the S. Cerevisiae genome (taken from SGD)
clustersencodes information about protein clusters; the proteins were clustered together based on their sequence similarity. (The details are not important.)A cluster begins with the line:
>Cluster N
where
Nis the cluster ID. The contents of the cluster are given in the following lines, for instance:>Cluster 54 0 >YHL009W-A at 90.80% 1 >YHL009W-B at 100.00% 2 >YJL113W at 98.77% 3 >YJL114W at 97.35%
represents a cluster (with ID
54) of four sequences. Four proteins belong to the cluster: protein"YHL009W-A"(with90.80%similarity to the cluster center), protein"YHL009-B"(with100.00%similarity) etc.Given the string
clusters, use a list comprehension (together with other operations on strings, such assplit()) for:Extracting the IDs of the various clusters. The expected result is:
>>> print cluster_ids ["0", "50", "54", "52"]
Extracting the names of all proteins, including duplicates. The expected result is:
>>> print protein_names ["YLR1106C", "YPL082C", "YHL00W-A", ...]
Extracting all protein-similarity pairs for all proteins. Th expected result is:
>>> print protein_similarity_pairs [["YLR106C", 100.0], ["YPL082C", 100.0], ["YHL009W-A", 90.8], # ... ]
Given the \(3\times 3\) matrix (list of lists):
matrix = [range(0,3), range(3,6), range(6,9)]
- Create a matrix
upside_downcontaining all rows ofmatrixfrom bottom to top. - (Hard.) Create a matrix
palindromecontaining all columns ofmatrixfrom left to right. - (Hard.) Re-create
matrixfrom scratch using a list comprehension.
- Create a matrix
(Hard.) Given the list:
list = range(100)
Create a list
squarescontaining the squares of all the elements inlist. The expected result is:[0, 1, 4, 9, ...]
Next, create a list
difference_of_squarescontaining, in every positioni, the value of:squares[i+1] - squares[i]
making sure to avoid the case
i == len(list)(because in that casesquares[i+1]is undefined).Feel free to use auxiliary variables as required.
(Blame this page for inspiring this exercise.)
Given the following list of mouse gene symbols:
mouse_genes = ["Fus", "Tdp43", "Sod1", "Ighmbp2", "Srsf2"]
- Sort the list alphabetically.
- In the sorted ist, convert mouse symbols into human gene symbols.
Hint: in human gene symbols all letters are in upper-case.
Python: Lists (Solutions)¶
Note
In some cases I will use the character \ at the end of a line.
Used in this way, \ tells Python that the command continues in the following line. Didn’t I use \, Python could think that the command is complete, giving an error message if the syntax is wrong.
You can ignore these \.
Operations¶
Solution:
list = [] print list, len(list) # check
Solution:
list = range(5) print list, len(list) # check print len(list)
Solution:
list = [0] * 100 print list, len(list) # check
Solution:
list_1 = range(10) list_2 = range(10, 20) list_complete = list_1 + list_2 print list_complete print list_complete == range(20) # True
Solution:
list = ["I am", "a", "list"] print list, len(list) # check print len(list[0]) print len(list[1]) print len(list[2])
Solution:
list = [0.0, "b", [3], [4, 5]] print len(list) # 4 print type(list[0]) # float print list[1], len(list[1]) # "b", 1 print list[2], len(list[2]) # [3], 1 print list[-1], len(list[-1]) # [4, 5], 2 print "b" in list # True print 4 in list # False print 4 in list[-1] # True
Solution: the first is a list of integers, the second a list of strings, the third is a string!:
print type(list_1) # list print type(list_2) # list print type(list_3) # str
Solutions:
# an empty list list = [] print len(list) # 0 del list # invalid syntax, Python gives an error message list = [} # a list that contains an empty list list = [[]] print len(list) # 1 print len(list[0]) # 0 del list # the following doesn't work because the list is not defined! list.append(0) # this works list = [] list.append(0) print list # [0] del list # this doesn't work because we forgot to put commas! list = [1 2 3] # this gives an error message because the list has only 3 elements! list = range(3) print list[3] # Extract the last element list = range(3) print list[-1] del list # Extract the first two elements (list[2], the third, # is excluded) list = range(3) sublist = list[0:2] print list del list # Extract all the elements(list[3], not existent # is excluded) list = range(3) sublist = list[0:3] print list del list # Extract the first two elements (list[-1], the third, # is excluded) list = range(3) sublist = list[0:-1] print list del list # Insert in third position the string "two" list = range(3) list[2] = "two" print list del list # this doesn't work, the list contains only three elements, # there is no fourth position, and Python gives an error list = range(3) list[3] = "three" # insert in third posizion the string "three" list = range(3) list[-1] = "three" print list del list # the index has to be an integer, Python gives an error message list = range(3) list[1.2] = "one point two" # substitute the second element of list (i.e. 1) # with a list of two strings; this is can be done, # since lists *can* contain other lists list = range(3) list[1] = ["protein1", "protein2"] print list del list
Solution:
matrix = [ [1, 2, 3], [4, 5, 6], [7, 8, 9], ] first_row = matrix[0] print first_row second_element_first_row = first_row[1] # or second_element_first_row = matrix[0][1] print second_element_first_row sum_first_row = matrix[0][0] + matrix[0][1] + matrix[0][2] print sum_first_row second_column = [matrix[0][1], matrix[1][1], matrix[2][1]] print second_column diagonal = [matrix[0][0], matrix[1][1], matrix[2][2]] print diagonal three_rows_together = matrix[0] + matrix[1] + matrix[2] print three_rows_together
Methods¶
First, let’s create a list. For example, an empty list:
list = []
next, let’s add the required elements with
append():list.append(0) list.append("text") list.append([0, 1, 2, 3])
Solution:
# add one 3 at the end of the list list = range(3) list.append(3) print list del list # add a list with a 3 at the end of the list list = range(3) list.append([3]) print list del list # add a 3 (the only element contained in the list [3]) # at the end of the list list = range(3) list.extend([3]) print list del list # doesn't work: extend() extends a list with the content of # another list, but here 3 is *not* a list! # Python gives an error message list = range(3) list.extend(3) # replace the element in position 0, the first, with a 3 list = range(3) list.insert(0, 3) print list del list # insert a 3 at the end of list list = range(3) list.insert(3, 3) print list del list # insert the list [3] at the end of list list = range(3) list.insert(3, [3]) print list del list # doesn't work: the first argument of insert() has to be an integer # not a list! Python gives an error message list = range(3) list.insert([3], 3)
Solution:
list = [] list.append(range(10)) list.append(range(10, 20)) print list
Here we use
append(), that inserts an element at the end oflist. In this example we insert two lists, the results ofrange(10)andrange(10, 20).Clearly,
len(list)is2, since we added only 2 elements.On the other hand:
list = [] list.extend(range(10)) list.extend(range(10, 20)) print list
here we use
extend(), that extends a list with another list. Here the final list has 20 elements, as we can see with:print len(list)
Solution:
list = [0, 0, 0, 0] list.remove(0) print list
only the first occurrence of
0is removed!Solution:
list = [1, 2, 3, 4, 5] # invert the order of the elements of list list.reverse() print list # order the elements of list list.sort() print list
After the two operations,``list`` gets back to the initial value.
On the other hand:
list = [1, 2, 3, 4, 5] list.reverse().sort()
cannot be done, since the result of
list.reverse()isNone:list = [1, 2, 3, 4, 5] result = list.reverse() print result
being result not a
list,``sort()`` cannot be applied. Python gives an error message.Let’s try this:
list = range(10) inverse_list = list.reverse() print list # modified! print inverse_list # None!
the code doesn’t work:
reverse()modifiesliste returnsNone! Moreover, this code modifies directly``list``, and we don’t want that.First, let’s create a copy of
list, next we can order the copy:list = range(10) inverse_list = list[:] # *not* inverse_list = list inverse_list.reverse() print list # unvaried print inverse_list # inverted
On the other hand, this code:
list = range(10) inverse_list = list inverse_list.reverse() print list # modified! print inverse_list # inverted
doesn’t work as we want:
inverse_listdoesn’t contain a copy oflist, but a reference to the same object referred bylist.As a consequence, when we invert
inverse_listwe also invertlist.As before:
motifs = [ "KSYK", "SVALVV" "GVTGI", "VGSSLAEVLKLPD", ] sorted_motifs = motifs.sort()
the code doesn’t work:
reverse()modifiesmotifse returnsNone! First, let’s create a copy ofmotifs, next we can order the copy:sorted_motifs = motifs[:] fsorted_motifs.sort() print motifs # unvaried print sorted_motifs # ordered
String-List Methods¶
Solution:
text = """The Wellcome Trust Sanger Institute is a world leader in genome research."""" words = text.split() print len(words)
Solution:
table = [ "protein | database | domain | start | end", "YNL275W | Pfam | PF00955 | 236 | 498", "YHR065C | SMART | SM00490 | 335 | 416", "YKL053C-A | Pfam | PF05254 | 5 | 72", "YOR349W | PANTHER | 353 | 414", ] first_row = table[0] almost_column_titles = first_row.split("|") almost_column_titles # ["protein ", " database ", ...] # unfortunately, column titles contain superfluous spaces # to remove them, we can change the argument of split() column_titles = first_row.split(" | ") print column_titles # ["protein", "database", ...]
We could also use
strip()together with a list comprehension onalmost_column_titles, but it’s not necessary.Solution:
words = ["word_1", "word_2", "word_3"] print " ".join(words) print ",".join(words) print " e ".join(words) print "".join(words) backslash = r"\" print backslash.join(words)
Solution:
verses = [ "Taci. Su le soglie" "del bosco non odo" "parole che dici" "umane; ma odo" "parole piu' nuove" "che parlano gocciole e foglie" "lontane." ] poem = "\n".join(verses)
List Comprehension¶
Solutions:
Solution:
list_plus_three = [number + 3 for number in list] print list_plus_three # check
Solution:
odds = [number for number in list if (number % 2 == 1)]
Solution:
opposites = [-number for number in list]
Solution:
inverses = [1.0 / number for number in list if number != 0]
Solution:
first_and_last = [list[0], list[-1]]
Solution:
from_second_to_penultimate = list[1:-1]
Solution:
list_odds = [number for number in list if (number % 2 == 1)] number_odds = len(list_odds) print number_odds
or:
number_odds = len([number for number in list if (number % 2 == 1)])
Solution:
list_divided_by_5 = [float(number) / 5 for number in list]
Solution:
list_multiples_5_divided = [float(number) / 5.0) for number in list if (number % 5 == 0)]
Solution:
list_of_strings = [str(number) for number in list]
Solution:
# As before, but iterating on `list_of_strings` # rather than directly on `list` number_odds = len([string for string in list_of_strings if (int(string) % 5 == 0)])
Solution:
text = " ".join([str(number) for number in list])
Notice that if we forget to write``str(number)``,
join()doesn’t work.
Solutions:
# forward list_1 = [1, 2, 3] list_2 = [str(x) for x in list_1] # backward list_2 = ["1", "2", "3"] list_1 = [int(x) for x in list_2]
# forward list_1 = ["name", "surname", "age"] list_2 = [[x] for x in list_1] # backward list_2 = [["name"], ["surname"], ["age"]] list_1 = [l[0] for l in list_2]
# forward list_1 = ["ACTC", "TTTGGG", "CT"] list_2 = [[x.lower(), len(x)] for x in list_1] # backward list_2 = [["actc", 4], ["tttgggcc", 6], ["ct", 2]] list_1 = [l[0].upper() for l in list_2]
Solution:
[x for x in list]: creates a copy oflist.[y for y in list]: creates a copy oflist(the same as before).[y for x in list]: invalid. (Ifxrepresents an element of the list, what isy?)["x" for x in list]: creates a list full of strings"x"as long aslist. The result will be:["x", "x", ..., "x"].[str(x) for x in list]: for each intxinlist,xis converted to a stringstr(x)and included in the resulting list. The result will be:["0", "1", ..., "9"].[x for str(x) in list]: invalid: the transformationstr(...)is in the wrong place![x + 1 for x in list]: for each intxinlist, adds 1 withx + 1and includes the result in the resulting list. The result will be:[1, 2, ..., 10].[x + 1 for x in list if x == 2]: for each intxinlistchecks whether it is equal to2. In the positive casex + 1is included in the resulting list, otherwise it’s excluded. The result will be’:[3].
Solution:
clusters = """\ >Cluster 0 0 >YLR106C at 100.00% >Cluster 50 0 >YPL082C at 100.00% >Cluster 54 0 >YHL009W-A at 90.80% 1 >YHL009W-B at 100.00% 2 >YJL113W at 98.77% 3 >YJL114W at 97.35% >Cluster 52 0 >YBR208C at 100.00% """ rows = clusters.split("\n")
In order to get cluster names, we have to keep only lines starting with
">", and for each of them applysplit()in order to get the second element (the nam of the cluster):cluster_names = [row.split()[1] for row in rows if row.startswith(">")]
In order to get protein names, we have to keep only lines not starting with
">", and for each of them applysplit()and keep the second element (also removing">"from the name of the protein):proteins = [row.split()[1].lstrip(">") for row in rows if not row.startswith(">")]
In order to get protein-percentage pairs, we have to keep only lines not starting with
">". On each of them, we applysplit()and keep the second element (protein name) and the last (percentage):protein_percentage_pairs = \ [[row.split()[1].lstrip(">"), row.split()[-1].rstrip("%")] for row in rows if not row.startswith(">")]
Annotated version:
protein_percentage_pairs = \ [[row.split()[1].lstrip(">"), row.split()[-1].rstrip("%")] # ^^^^^^^^^^^^^^^^^^^^^^^^^^^ ^^^^^^^^^^^^^^^^^^^^^^^^^^^^ # protein name, as before percentage # ^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ # protein-percentage pair for row in rows if not row.startswith(">")]
Solution:
matrix = [range(0,3), range(3,6), range(6,9)] # extract first row first_row = matrix[0] # extract first column first_column = [matrix[0][i] for i in range(3)] # invert row order upside_down = matrix[:] upside_down.reverse() # or upside_down = [matrix[2-i] for i in range(3)] # invert column order palindrome = [] # append the first line palindrome.append([matrix[0][2-i] for i in range(3)]) # append the second line palindrome.append([matrix[1][2-i] for i in range(3)]) # append the third line palindrome.append([matrix[2][2-i] for i in range(3)]) # or in a single step -- it's complicated and you can ignore it!!! palindrome = [[row[2-i] for i in range(3)] for row in matrix] # we can re-create matrix with a single list comprehension matrix_again = [range(i, i+3) for i in range(9) if i % 3 == 0]
Solutions:
list = range(100) squares = [number**2 for number in list] difference_of_squares = \ [squares[i+1] - squares[i] for i in range(len(squares) - 1)]
Solutions:
mouse_genes = ["Fus", "Tdp43", "Sod1", "Ighmbp2", "Srsf2"] sorted_mouse_genes = mouse_genes[:] sorted_mouse_genes.sort() print sorted_mouse_genes human_genes = [gene.upper() for gene in sorted_mouse_genes]
Python: Tuple¶
Tuples are immutable lists.
In order to define a tuple, use this syntax:
binding_partners = (
"BIOGRID:112315",
"BIOGRID:108607" # no comma
)
Leaving an additional comma after the last element also works:
binding_partners = (
"BIOGRID:112315",
"BIOGRID:108607", # this comma makes no difference
)
Tuples should be used instead of lists whenever mutability is an issue, for instance as dictionary keys.
Example. Let’s create a bunch of different tuples:
# A tuple of integers
integers = (1, 2, 3, 1)
# A tuple of strings
uniprot_proteins = ("Y08501", "Q95747")
# A tuple of heterogeneous objects
things = ("Y08501", 0.13, "Q95747", 0.96)
# A tuple of tuples
two_level_tuple = (
("Y08501", 120, 520),
("Q95747", 550, 920),
)
Example. Empty and length-one tuples are a bit weird. I will not explain how to create empty tuples, they are very rare (and not really useful).
But let’s assume for the sake of illustration that we need to create
a tuple with just one element, say the integer 0. This does not work:
tuple = (0)
print tuple
print type(tuple) # not a tuple!
The reason is that (0) is interpreted by Python as an arithmetical
expression. Indeed, this is what the syntax means: an integer within a pair of
brackets.
In order to define such a one-element tuple, write:
tuple = (0,)
print tuple
print type(tuple) # it's a tuple!
Now the ambiguity is gone: the comma , in there makes it clear to Python
that we want to create a tuple – except that it contains only one element.
Operators¶
Most list operators can be used with tuples as well. Of course, since tuples are immutable, operators that change the value of lists (like the assignment operator) do not carry over to tuples.
| Returns | Operator | Meaning |
|---|---|---|
int |
len(tuple) |
Compute the length of a tuple |
tuple |
tuple + tuple |
Concatenate two tuples |
tuple |
tuple * int |
Replicate a tuple |
bool |
object in tuple |
Check whether an element appears in a tuple |
tuple |
tuple[int:int] |
Extract a sub-tuple |
Example. Let’s create a tuple with mixed object types:
protein_info = ("2B0Q", "phosphotransferase", "PF03881")
It holds information about a given protein (PDB identifier 2B0Q). We can access the various elements as if it were a list:
protein_id = protein_info[0]
print protein_id
protein_name = protein_info[1]
print protein_name
However, we can not modify the tuple in any way:
>>> protein_info[0] = "1A3A"
Traceback (most recent call last):
File "<stdin>", line 1, in <module>
TypeError: 'tuple' object does not support item assignment
Methods¶
| Returns | Method | Meaning |
|---|---|---|
int |
list.index(object) |
Return the index of the first occurrence of an object |
int |
list.count(object) |
Count the number of occurrences of an object |
These are identical to their list counterparts.
List-Tuple Conversion¶
In some cases you want to modify a tuple, but you can’t. In other cases you want to use a list as a key in a dictionary, but you can’t.
The tricks is to convert between tuples and lists as needed. In order to
convert between the two, use the tuple() and list() functions:
l = range(10)
print l, type(l)
t = tuple(l)
print t, type(t)
l = list(t)
print l, type(l)
Example. We want to modify a given tuple:
t = ("I", "am", "a", "tuple")
print t
Let’s proceed as follows:
l = list(t)
l.insert(2, "pretty")
t2 = tuple(l)
print t
print t2
Of course, we did not modify the original tuple; rather, we created a new tuple from an intermediate list.
Exercises¶
Create:
A tuple with two integers,
0and1.A tuple with two strings,
"a"and"tuple".A tuple with only one element,
"a tuple".Hint: you can either use the syntax presented at the beginning of the chapter, or use a list-tuple conversion.
A tuple with the integers between 0 and 99.
Hint: is there some way to exploit the
range()function?
Given
l = [0, 1, 2]andt = (0, 1, 2), what is the difference between:First case:
x = l x[0] = "ok"
Second case:
x = t x[0] = "ok"
Given the tuple:
tup = (0, 1, 2, [3, 4, 5], 6, 7, 8)
- What is the type of the first element of the tuple?
- What is the type of the fourth element?
- How many elements does the tuple contain?
- How many elements does the inner list contain?
- Change the last element of the inner list to
"last element". - Change the last element of the tuple to
"very last element". (Can you do that without resorting to tricks?)
Python: Tuple (Solutions)¶
Solutions:
couple_of_int = (0, 1) print type(couple_of_int) # tuple couple_of_strings = ("one", "tuple") print type(couple_of_strings) # tuple a_single_element = (0,) print type(a_single_element) # tuple print len(a_single_element) # 1 a_single_element_alt = tuple([0]) print type(a_single_element_alt) # tuple print len(a_single_element_alt) # 1 wrong = (0) print type(wrong) # int print len(wrong) # error! hundred_elements = tuple(range(100)) print type(hundred_elements) # tuple tuple_of_lists = (range(50), range(50, 100)) print type(tuple_of_lists) print type(tuple_of_lists[0]) tuple_of_tuples = (tuple(range(50)), tuple(range(50, 100))) print type(tuple_of_tuples) print type(tuple_of_tuples[0])
Solutions:
l = [0, 1, 2] t = (0, 1, 2) # x refers to a list, the code replaces # the first element with 100 x = l x[0] = 100 # x now refers to a tuple, that is immutable: # we cannot replace its elements, Python raises an error message x = t x[0] = 100 # error!
Solutions:
tup = (0, 1, 2, [3, 4, 5], 6, 7, 8) print tup[0] # 0 print type(tup[0]) # int print tup[3] # [3, 4, 5] print type(tup[3]) # list print len(tup) # 9 print len(tup[3]) # 3 tup[3][-1] = "last" print tup # yes, we can do it, we "modified" the tuple # by modifying the list contained in it. tup[-1] = "last" # error! # we cannot modify the tuple "directly" # it's an immutable object
Python: Dictionaries¶
A dictionary represents a map between objects: it maps from a key to the corresponding value.
Warning
Just like lists, dictionaries are mutable!
The abstract syntax for defining a dictionary is:
{ key1: value1, key2: value2, ... }
Example. In order to define a dictionary implementing the genetic code, we write:
genetic_code = {
"UUU": "F", # phenilalanyne
"UCU": "S", # serine
"UAU": "Y", # tyrosine
"UGU": "C", # cysteine
"UUC": "F", # phenilalanyne
"UCC": "S", # serine
"UAC": "Y", # tyrosine
# etc.
}
Here genetic_code maps from three-letter nucleotide strings (the keys) to
the corresponding amino acid character (the value).
To use a dictionary, we resort to the usual extraction operator, as follows:
>>> aa = genetic_code["UUU"]
>>> print aa
"phenylalanine"
>>> aa = genetic_code["UCU"]
>>> print aa
"serine"
I can use the genetic_code dictionary to “simulate” the process of translation and convert an RNA sequence into the corresponding aminoacid sequence. For instance, starting from the following mRNA sequence:
rna = "UUUUCUUAUUGUUUCUCC"
I can split it in triples:
>>> triples = [rna[i:i+3] for i in range(0, len(rna), 3)]
>>> print triples
['UUU', 'UCU', 'UAU', 'UGU', 'UUC', 'UCC']
At this point, I can translate the triples with:
>>> aminoacids = [genetic_code[triple] for triple in triples]
>>> protein = "".join(aminoacids)
>>> print protein
"FSYCFS"
Of course, this is a very simple functional model of translation. The most obvious difference is that the above Python code does not care about termination codons. Support for them will be added in due time.
Warning
Keys are unique: the same key can not be used more than once.
Value are not unique: different keys can map to the same value.
In the genetic code example, each key is a unique three-letter string,
which is associated to a given value; the same value (for instance "serine"
is associated to multiple keys:
print genetic_code["UCU"] # "S", serine
print genetic_code["UCC"] # "S", serine
Example. Let’s build a dictionary that maps from amino acids to their (approximated) volume in cubic Amstrongs:
volume_of = {
"A": 67.0, "C": 86.0, "D": 91.0,
"E": 109.0, "F": 135.0, "G": 48.0,
"H": 118.0, "I": 124.0, "K": 135.0,
"L": 124.0, "M": 124.0, "N": 96.0,
"P": 90.0, "Q": 114.0, "R": 148.0,
"S": 73.0, "T": 93.0, "V": 105.0,
"W": 163.0, "Y": 141.0,
}
# Print the volume of a cysteine
print volume_of["C"] # 86.0
print type(volume_of["C"]) # float
Here the keys are strings and the values are floats.
Warning
There are no restrictions on the kinds of objects that can appear as values.
The keys, however, must be immutable objects. This means that list
and dict objects can not be used as keys.
More formally, here is what object types you can use where:
| Type | As keys | As values |
|---|---|---|
| bool | ✓ | ✓ |
| int | ✓ | ✓ |
| float | ✓ | ✓ |
| str | ✓ | ✓ |
| list | NO | ✓ |
| tuple | ✓ | ✓ |
| dict | NO | ✓ |
This restriction is due to how dictionaries are implemented in Python (and most other programming languages, really).
Example. Let’s create a dictionary that maps from amino acids to a list of two properties, mass and volume:
properties_of = {
"A": [ 89.09, 67.0],
"C": [121.15, 86.0],
"D": [133.10, 91.0],
"E": [147.13, 109.0],
# ...
}
# Print the properties of alanine
print properties_of["A"] # [89.09, 67.0]
print type(properties_of["A"]) # list
Here the keys are str (immutable) and the values are list (mutable).
Can I create the inverse dictionary (from property list to amino acids)?
No. Let’s write the very first key-value pair:
aa_of = { [89.09, 67.0]: "A" }
Ideally, this dictionary would map from the list:
[89.09, 67.0]
to "A". However Python raises an error:
Traceback (most recent call last):
File "<stdin>", line 1, in <module>
TypeError: unhashable type: 'list'
# ^^^^^^^^^^^^^^^^^^^^^^^
# read: list is mutable!
To solve the problem, I can use tuples in place of lists:
aa_of = {
( 89.09, 67.0): "A",
(121.15, 86.0): "C",
(133.10, 91.0): "D",
(147.13, 109.0): "E",
# ...
}
aa = aa_of[(133.10, 91.0)]
print aa # "D"
print type(aa) # str
Now that the keys are immutable, everything works.
Operations¶
| Returns | Operator | Meaning |
|---|---|---|
int |
len(dict) |
Return the number of key-value pairs |
object |
dict[object] |
Extracts the value associated to a key |
| – | dict[object]=object |
Inserts or replaces a key-value pair |
The only major behavioral difference lies in the assignment operator (the last one). The syntax is the same as for lists, and the meaning too; however, for dictionaries it can be used to add entirely new key-value pairs. Let’s see a few examples.
Example. Starting from an empty dictionary:
code = {}
print code
print len(code)
I want to build (this time, incrementally) the genetic code dictionary I introduced in the very first example of this chapter. Let’s add the key-value pairs one by one with the assignment operator:
code["UUU"] = "F" # phenylalanine
code["UCU"] = "M" # methionine
code["UAU"] = "Y" # tyrosine
# ...
print code
print len(code)
Here I am adding new key-value pairs to a dictionary.
Whoops, I made a mistake! "UCU" should map to an "S", not to an
"M"! I can solve the problem by replacing the value associated to the key
"UCU", using the same syntax as above:
code["UCU"] = "S" # serine
print code
print len(code)
So, if the key was already there, the assignment operator simply replaces the value it is associated with.
Qui alla key "UCU", che gia’ era nel dizionario, associo un nuovo value
"S" (serina).
Warning
It does not make sense, however, to extract values associated to keys not present in the dictionary. For instance:
>>> code[":-("]
makes Python raise an error:
Traceback (most recent call last):
File "<stdin>", line 1, in <module>
KeyError: ":-("
# ^^^^^
# the dictionary contains no such key
Methods¶
| Returns | Method | Meaning |
|---|---|---|
bool |
dict.has_key(object) |
True if the object is a key of the dict |
list |
dict.keys() |
Get the keys as a list |
list |
dict.values() |
Get the values as a list |
list-of-tuples |
dict.items() |
Get the key-value pairs as a list of pairs |
Example. Starting from code:
code = {
"UUU": "F", # phenylalanine
"UCU": "S", # serine
"UAU": "Y", # tyrosine
"UGU": "C", # cysteine
"UUC": "F", # phenylalanine
"UCC": "S", # serine
"UAC": "Y", # tirosine
# ...
}
I can get the list of keys:
>>> keys = code.keys()
>>> print keys
["UUU", "UCU", "UAU", ...]
and the list of values:
>>> values = code.values()
>>> print values
["F", "S", Y", "C", ...]
and the list of both keys and values:
>>> key_value_pairs = code.items()
>>> print key_value_pairs
[("UUU", "F"), ("UCU", "S"), ...]
Now that I have a bunch of lists, I can apply any of the list operations/methods to perform the tasks I need!
Finally, to check whether a given object appears as a key, I can write:
>>> print code.has_key("UUU")
True
>>> print code.has_key(":-(")
False
Warning
Key-value pairs are stored in the dictionary in a seemingly arbitrary order.
In other words, Python does not guarantee that the order in which the key-value pairs are inserted is preserved.
For instance:
>>> d = {}
>>> d["z"] = "zeta"
>>> d["a"] = "a"
>>> d
{'a': 'a', 'z': 'zeta'}
>>> d.keys()
['a', 'z']
>>> d.values()
['a', 'zeta']
>>> d.items()
[('a', 'a'), ('z', 'zeta')]
Here I inserted ("z", "zeta") first, and ("a", "a") second.
However, the order in which they are stored in the dictionary is the
exact opposite!
Example. We can use a dictionary to represent a complex structured object, for instance the properties of a protein chain:
chain = {
"name": "1A3A",
"chain": "B",
"sequence": "MANLFKLGAENIFLGRKAATK...",
"num_scop_domains": 4,
"num_pfam_domains": 1,
}
print chain["name"]
print chain["sequence"]
print chain["num_scop_domains"]
Of course, writing a dictionary like this by hand is inconvenient. We will later see how such dictionaries can be created by reading the data automatically out of some biological database.
(Imagine having 100k such dictionaries to analyze!)
Example. Given the following FASTA sequence (cut down to a reasonable size) describing the primary sequence of the HIV-1 retrotranscriptase protein (taken from the PDB):
>2HMI:A|PDBID|CHAIN|SEQUENCE
PISPIETVPVKLKPGMDGPKVKQWPLTEEKIKALVEICTEMEKEGKISKI
NKRTQDFWEVQLGIPHPAGLKKKKSVTVLDVGDAYFSVPLDEDFRKYTAF
QSSMTKILEPFKKQNPDIVIYQYMDDLYVGSDLEIGQHRTKIEELRQHLL
VQPIVLPEKDSWTVNDIQKLVGKLNWASQIYPGIKVRQLSKLLRGTKALT
PSKDLIAEIQKQGQGQWTYQIYQEPFKNLKTGKYARMRGAHTNDVKQLTE
WWTEYWQATWIPEWEFVNTPPLVKLWYQLEKEPIVGAETFYVDGAANRET
AIYLALQDSGLEVNIVTDSQYALGIIQAQPDKSESELVNQIIEQLIKKEK
>2HMI:B|PDBID|CHAIN|SEQUENCE
PISPIETVPVKLKPGMDGPKVKQWPLTEEKIKALVEICTEMEKEGKISKI
NKRTQDFWEVQLGIPHPAGLKKKKSVTVLDVGDAYFSVPLDEDFRKYTAF
QSSMTKILEPFKKQNPDIVIYQYMDDLYVGSDLEIGQHRTKIEELRQHLL
VQPIVLPEKDSWTVNDIQKLVGKLNWASQIYPGIKVRQLSKLLRGTKALT
PSKDLIAEIQKQGQGQWTYQIYQEPFKNLKTGKYARMRGAHTNDVKQLTE
WWTEYWQATWIPEWEFVNTPPLVKLWYQLE
>2HMI:C|PDBID|CHAIN|SEQUENCE
DIQMTQTTSSLSASLGDRVTISCSASQDISSYLNWYQQKPEGTVKLLIYY
EDFATYYCQQYSKFPWTFGGGTKLEIKRADAAPTVSIFPPSSEQLTSGGA
NSWTDQDSKDSTYSMSSTLTLTADEYEAANSYTCAATHKTSTSPIVKSFN
>2HMI:D|PDBID|CHAIN|SEQUENCE
QITLKESGPGIVQPSQPFRLTCTFSGFSLSTSGIGVTWIRQPSGKGLEWL
FLNMMTVETADTAIYYCAQSAITSVTDSAMDHWGQGTSVTVSSAATTPPS
TVTWNSGSLSSGVHTFPAVLQSDLYTLSSSVTVPSSTWPSETVTCNVAHP
>2HMI:E|PDBID|CHAIN|SEQUENCE
ATGGCGCCCGAACAGGGAC
>2HMI:F|PDBID|CHAIN|SEQUENCE
GTCCCTGTTCGGGCGCCA
We’d like to compile a dictionary with the same information:
sequences_2HMI = {
"A": "PISPIETVPVKLKPGMDGPKVKQWPLTEEKI...",
"B": "PISPIETVPVKLKPGMDGPKVKQWPLTEEKI...",
"C": "DIQMTQTTSSLSASLGDRVTISCSASQDISS...",
"D": "QITLKESGPGIVQPSQPFRLTCTFSGFSLST...",
"E": "ATGGCGCCCGAACAGGGAC",
"F": "GTCCCTGTTCGGGCGCCA",
}
From this dictionary, it’s very easy to extract the sequence of every individual chain:
>>> print sequences_2HMI["F"]
"GTCCCTGTTCGGGCGCCA"
as well as computing how many chains there are:
num_catene = len(sequences_2HMI)
Example. Dictionaries can be used to describe histograms. For instance, let’s take a sequence:
seq = "GTCCCTGTTCGGGCGCCA"
We compute the number of the various nucleotides:
num_A = seq.count("A") # 1
num_T = seq.count("T") # 4
num_C = seq.count("C") # 7
num_G = seq.count("G") # 6
It is easy to write down a corresponding histogram into a dictionary:
histogram = {
"A": float(num_A) / len(seq), # 1 / 18 ~ 0.06
"T": float(num_T) / len(seq), # 4 / 18 ~ 0.22
"C": float(num_C) / len(seq), # 7 / 18 ~ 0.38
"G": float(num_G) / len(seq), # 6 / 18 ~ 0.33
}
Let’s say we are now interested in the proportion of adenosine; we can write:
prop_A = histogram["A"]
print prop
We can also check whether the histogram represents a “true” multinomial distribution by checking whether the sum of the probabilities is (approximately) 1:
print histogram["A"] + histogram["C"] + ...
Example. Dictionaries are also very useful for describing protein interaction networks (physical, functional, genomic, you name it):
partners_of = {
"2JWD": ("1A3A",),
"1A3A": ("2JWD", "3BLU", "2ZTI"),
"2ZTI": ("1A3A", "3BLF"),
"3BLU": ("1A3A", "3BLF"),
"3BLF": ("3BLU", "2ZTI"),
}
which represents the following network:
2JWD ----- 1A3A ----- 2ZTI
| |
| |
3BLU ----- 3BLF
Here partners_of["1A3A"] is a tuple with all of the binding partners of
protein 1A3A.
We can use the dictionary to compute the n-step neighborhood of 1A3A:
# 1-step neighborhood of 1A3A
neighborhood_at_1 = partners_of["1A3A"]
# 2-step neighborhood of 1A3A (it may include repeated elements)
neighborhood_at_2 = \
[partners_of[p] for p in neighborhood_at_1]
# 3-step neighborhood of 1A3A (again, may include repeated elements)
neighborhood_at_3 = \
[partners_of[p] for p in neighborhood_at_2]
Note that the very same idea can be used to encode social networks (Facebook, Twitter, Google+), to see who-is-friends-with-who, and to discover communities in said networks.
Note
Dictionaries are just one of the many ways to encode a network (or graph). An alternative is to use an adjacency matrix, which can be implemented (as usual) as a list of lists.
Exercises¶
Create manually:
An empty dictionary. Check whether the dictionary is empty with
len().A dictionary
pronounsrepresenting these correspondences:1 -> "me" 2 -> "you" 3 -> "he" ...
Create the dictionary in 2 ways:
- In a single line of code.
- Starting from an empty dictionary and adding one by one single key value pairs.
A dictionary
tenfold_ofimplementing the function \(f(n) = 10 n\), matching each key (an integer) with its tenfold.Keys should be integers from 1 to 5.
Once the dictionary is built, apply it to all the elements of
range(2, 5)with a list comprehension and print to screen the result.Repeat the same for all the keys of
tenfold_of.Hint: you can obtain the list of all keys with
keys().A dictionary
info_2TMVcodifying structured information about the PDB entry 2TMV containing the structure of a capsid protein of the Tobacco Mosaic Virus.- To the key
"pdb_id"associate the value"2TMV". - To the key
"uniprot_id"associate the value"P69687 (CAPSD_TMV)". - To the key
"number_scp_domains"associate1. - To the key
"number_pfam_domains"associate1.
- To the key
A dictionary
relatives_ofrepresenting this network:JULIE ---- FRANCIS ---- MATTHEW | | + ----- BENEDICT -------+
as in the example we saw before.
Once the dictionary is built, print to screen the number of relatives of
"JULIE".A dictionary
from_2_bit_to_integerrepresenting the map from the following pairs of integers to integer:0, 0 -> 0 # 0*2**1 + 0*2**0 = 0 0, 1 -> 1 # 0*2**1 + 1*2**0 = 1 1, 0 -> 2 # 1*2**1 + 0*2**0 = 2 1, 1 -> 3 # 1*2**1 + 1*2**1 = 3 ^^^^ ^ ^^^^^^^^^^^^^^^^^^^ key value explanation
Once the dictionary is built, pront to screen the value associated with one of the keys (choose arbitrarily).
Given:
ratios = { ("A", "T"): 10.0 / 5.0, ("A", "C"): 10.0 / 7.0, ("A", "G"): 10.0 / 6.0, ("T", "C"): 5.0 / 7.0, ("T", "G"): 5.0 / 6.0, ("C", "G"): 7.0 / 6.0, }
representing ratios between the number of A, T, C, e G in a sequence:
What is the difference between
len(ratios),len(ratios.keys()),len(ratios.values())elen(ratios.items())?Check whether
ratioscontains the key("T", "A"). And the key("C", "G")?Hint: can we use
keys()? Can we use another method?Check whether it contains the value 2. And the value 3?
Hint: can we use
values()?Check whether it contains the key-value pair
(("A", "T"), 2). And the key-value pair(("C", "G"), 1000)?Hint: can we use
items()?Use a list comprehension to extract the keys of the result of
items(). Do the same to extract values.
Given:
map = { "zero": 1, "one": 2, "two": 4, "three": 8, "four": 16, "five": 32, }
Concatenate all the keys
map, separated by spaces, in a single stringstring_of_keys.Concatenate all the values of
mapas strings, separated by spaces, in a single stringstring_of_values.Hint: note that the values of
mapare not strings!Insert in a list all the keys of
map.Insert in a list all the keys of
map, ordered alphanumerically.Hint: is the list returned by
keys()ordered?Insert in a list all the values of
map, ordered according to the order of the corresponding keys.Hint: how can I order a list based on the order of another list?
Given:
translation_of = {"a": "ade", "c": "cyt", "g": "gua", "t": "tym"}
translate the list:
list = ["A", "T", "T", "A", "G", "T", "C"]
into the string:
"ade tym tym ade gua tym cyt"Hint: note that dictionary keys are in lower case, while the elements of
listare in upper case! Start assuming they are not, then modify the code in order to account for this difference.
Python: Dictionaries (Solutions)¶
Solutions:
Solution:
empty_dict = {} print empty_dict print len(empty_dict) # 0
Solution:
pronouns = {} pronouns[1] = "I" pronouns[2] = "you" pronouns[3] = "he" pronouns[4] = "we" pronouns[5] = "you" pronouns[6] = "they"
or:
pronouns = { 1: "I", 2: "you", 3: "he", 4: "we", 5: "you", 6: "them", }
Solution:
tenfold_of = {1: 10, 2: 20, 3: 30, 4: 40, 5: 50} print [tenfold_of[n] for n in range(2, 5)] print [tenfold_of[key] for key in tenfold_of.keys()]
Solution:
info_2TMV = { "pdb_id": "2TMV", "uniprot_id": "P69687 (CAPSD_TMV)", "number_scp_domains": 1, "number_pfam_domains": 1, }
Solution:
relatives_of = { "JULIE": ["FRANCIS", "BENEDICT"], "FRANCIS": ["JULIE", "MATTHEW"], "MATTHEW": ["FRANCIS", "BENEDICT"], "BENEDICT": ["JULIE", "MATTHEW"], } num_relatives_of_julie = len(relatives_of["JULIE"]) print num_relatives_of_julie
Solution:
from_2_bit_to_integer = { (0, 0): 0, (0, 1): 1, (1, 0): 2, (1, 1): 3, }
Note that we cannot use lists as keys: lists are not immutable!
Let’s print the value corresponding to 1, 0:
print from_2_bit_to_integer[(1, 0)] # ^^^^^^ # tuple
Solution:
ratios = { ("A", "T"): 10.0 / 3.0, ("A", "C"): 10.0 / 7.0, ("A", "G"): 10.0 / 6.0, ("T", "C"): 3.0 / 7.0, ("T", "G"): 3.0 / 6.0, ("C", "G"): 7.0 / 6.0, } print len(ratios) # 6 print len(ratios.keys()) # 6 print len(ratios.values()) # 6 print len(ratios.items()) # 6 # they all count the number of key-value pairs! # let's pront the keys of the dictionary, to get an idea print ratios.keys() # a list of tuples! print type(ratios.keys()) # list print type(ratios.keys()[0]) # tuple contains_T_A = ("T", "A") in ratios.keys() print contains_T_A # False contains_C_G = ("C", "G") in ratios.keys() print contains_C_G # True # let's do the same with has_key() print ratios.has_key(("T", "A")) # False print ratios.has_key(("C", "G")) # True # let's print the values of the dictionary to get an idea print ratios.values() # a list of int! print type(ratios.values()[0]) # int contains_2 = 2 in ratios.values() print contains_2 # True contains_3 = 3 in ratios.values() print contains_3 # False # let's print the key-value pairs to get an idea print ratios.items() # it's a list of pairs (tuple): the first element, the key, is # a tuple itself, the second is an int print (("A", "T"), 2) in ratios.items() # True print (("C", "G"), 1000) in ratios.items() # False # list comprehension: keys = [key_value[0] for key_value in ratios.items()] values = [key_value[-1] for key_value in ratios.items()]
Solution:
map = { "zero": 1, "one": 2, "two": 4, "three": 8, "four": 16, "five": 32, } # all the keys of map are strings so keys() returns a list of strings # we can use directly join() string_of_keys = " ".join(map.keys()) # the values of map are int, so we cannot use directly join() # we first have to transform all values from int to string string_of_values = " ".join(str(value) for value in map.values()) list_of_keys = map.keys() print list_of_keys # not ordered ordered_list_of_keys = map.keys() ordered_list_of_keys.sort() print ordered_list_of_keys # now it is ordered list_of_values_ordered_by_keys = \ [map[key] for key in ordered_list_of_keys]
Solution:
# using a list comprehension we can apply the dictionary to # the list: it's necessary to use lower() *before* # using an aminoacid as a key of translation_of! translation= [translation_of[aa.lower()] for aa in list] print translation # now we can use join() to concatenate the various parts result = " ".join(translation) print result
or, in a single line:
print " ".join([translation_of[aa.lower()] for aa in list])
Solution:
propensities = { 'N': 0.2299, 'P': 0.5523, 'Q': -0.1877, 'A': -0.2615, 'R': -0.1766, 'S': 0.1429, 'C': -0.01515, 'T': 0.0089, 'D': 0.2276, 'E': -0.2047, 'V': -0.3862, 'F': -0.2256, 'W': -0.2434, 'G': 0.4332, 'H': -0.0012, 'Y': -0.2075, 'I': -0.4222, 'K': -0.100092, 'L': 0.33793, 'M': -0.22590 } # using a list comprehension pos_aa = [aa for aa in propensities.keys() if propensities[aa]>0] print pos_aa
Python: Input-Output¶
Interacting with the User¶
The simplest kind of interaction with the user is to ask a question (given as a string) and read back the answer (also as a string).
The raw_input() function allows to do it.
| Returns | Function | Meaning |
|---|---|---|
str |
raw_input([str]) |
Asks a question to the user, returns the answer |
Example. Let’s ask the user a simple question:
answer = raw_input("write three space-separated words: ")
print answer
print type(answer)
Here the answer variable contains the user answer. It is always a
string; the user decides its actual value.
Now, let’s check if the user answered with exactly three words:
words = answer.split()
print "you wrote", len(words), "words"
reaction_to = {
True: "Nice!",
False: "You can do better than that."
}
print reaction_to[len(words) == 3]
Example. Let’s ask the user:
answer = raw_input("write two numbers: ")
print answer
print type(answer)
Let’s print the sum of the two numbers provided by the user. Note that, since
the value returned by raw_input() is always a string, we have to
first split into words, and then convert the words into (say) floats:
words = answer.split()
n = float(words[0])
m = float(words[1])
print "the sum is", n + m
Of course, if the user can not be split into two words, or if those words do not represent valid float values, the code above will not work.
Reading and Writing Text Files¶
In order to access the contents of a file (let’s assume a text file for
simplicity), we need to first create a handle to it. This can be done
with the open() function (see below).
Warning
A handle is simply an object that refers to a given file.
It does not contain any of the file data, but it can be used together with other methods, to read and write from the file it refers to.
| Returns | Function | Meaning |
|---|---|---|
file |
open(str, [str]) |
Get a handle to a file |
str |
file.read() |
Read all the file as a single string |
list-of-str |
file.readlines() |
Read all lines of the file as a list of strings |
str |
file.readline() |
Read one line of the file as a string |
None |
file.write(str) |
Write one string to the file |
| – | file.close() |
Close the file (= flushes changes to disk) |
open() allows to get a handle to a file.
The first argument to open() is the path of the file we want to open.
The second argument is optional. It tells open() how we intend to
use the file: for reading, for writing, etc.
These are the available access modes:
"r": we want to read from the file. This is the default mode."w": we want to write to the file, overwriting its contents."a": we want to write to the file, appending to the existing contents.
Mode flags can be combined. For instance:
"rw": we want to read and write (in “overwrite” mode)."ra": we want to read and write (in “append” mode).
Once you are done with a file (either reading or writing), make sure
to call the close() method to finalize your operations.
Example. Let’s open a file in read-only mode:
handle = open("data/aatable", "r")
print type(handle)
Once we have the file handle, we can read the contents of the file:
As a single, big string:
all_contents = handle.read() print all_contents
read()makes sense if your file is small enough (i.e. it fits into the RAM) and it is not structured as a sequence of lines separated by newline characters.Some files fit this description (they are pretty rare, tho).
As a list of lines (strings):
lines = handle.readlines() print lines
readlines()makes sense if your file is small enough and it is structured as a collection of lines.FASTA files are an example of such files.
One line at a time, sequentially, from the first onwards:
line = handle.readline() print line line = handle.readline() print line # etc.
readline()makes sense for very large files, because you can read one line at a time, without saturating the machine.
Given that the handle has been opened in read-only mode (that is, "r")
we can not write to it:
>>> handle.write("something something")
Traceback (most recent call last):
File "<stdin>", line 1, in <module>
IOError: File not open for writing
# ^^^^^^^^^^^^^^^^^^^^^^^^^
# Python complains about it!
Warning
Internally, Python keeps track of which lines of a file
have already been read. (This is used to implement the
readline() method, for instance.)
Once a line has been read, it can not be read from the same file handle.
This limitation affects all three methods: read(),
readlines() and readline().
For instance:
handle = open("data/aatable")
# No line has been read yet, so
lines = handle.readlines()
print len(lines)
# I just read *all* of the lines; no unread
# line is left
whats_left = handle.readlines()
print len(whats_left)
# Here Python did *not* read anything, because
# there were no lines left to read!
Example. Let’s see how to write to a file. We have to open a
file handle in write mode (either "w" or "a"):
# Open a file for writing
handle = open("result.txt", "w")
# TODO: write a long and complex calculation whose
# result is 42
result = 42
# In order to write the result (which is an int) to the
# file, we have to convert it to a string. Let's also
# add a newline character, so that the result fills a
# whole line.
handle.write(str(result))
# Make sure that our writes are written to disk.
handle.close()
And that’s it.
Warning
Forgetting to close a file opened in read-only mode is not too harmful.
(It is only harmful if you keep opening files and end up with more open files than the Operating System allows. Yes, there is a limit.)
However, forgetting to explicitly close files opened in
write mode (either "w", "a" or any other combination
including them) can have serious consequences.
The reason is that writes to files are not immediately
written to disk, for efficiency. Instead, they are stored
in memory until Python decides to write them to the actual
file. close() is a way to tell Python to flush the
changes to the file.
Intuitively, this means that if you don’t call close()
and the program quits (because of an error, for instance),
then your changes are not written to the file.
Example. We ask the user the path to a file (it does not matter whether it already exists or not), and then what to write in it:
path = raw_input("give me a path: ")
text = raw_input("give me the contents: ")
handle = open(path, "w")
handle.write("the user replied " + text)
handle.close()
Quiz: what happens if you execute this code twice on the same file?
Now, let’s open the same file, but for reading, and read its contents:
other_handle = open(path, "r")
all_contents = other_handle.read()
other_handle.close()
print "I found:"
print all_contents
print "in the file at", path
Exercises (raw input)¶
Using
raw_input(), ask the user for his/her favourite food, store the result in a variablefood, then print to screen:I also likeand the favourite food.Ask the user for two integers, let’s say
aandb, then a third integer, let’s sayresult.Check whether the sum of
aandbis equal toresult: if so, print to screenTrue, otherwiseFalse.Ask the user for a key and a value, and build a dictionary including (only) that key-value pair. Print to screen the result.
Ask the user for his/her name, store it in the variable
name, then print to screen the name, making sure that all the words contained innameare in lower case, except for the first characters, that must be in upper case.
Exercises (Filesystem)¶
Warning
If open() raises an error, maybe you are in the wrong directory: fix the path in order to be in the right directory.
Use
open()to open the file"data/aatable"in read-only mode, storing the result ofopen()in the variablef.Next, use
readlines()to read the content of the file, storing the result inrows.What is the type of
rows? What is the type of the first element ofrows? How many rows are there in the file?Use
open()to open the file"data/aatable"in read-only mode, storing the result ofopen()in the variablef.Next, use
readline()(notreadlines()!) to read the first row of the file, storing the result infirst_row.How many rows are left to read? Check using
readlines().At this point, how many other rows are left to read?
Use
open()to read the file"output.txt"in write-only mode"w", storing the result ofopen()in the variablef.Next, write the string
"check one two three check"in the file.Close the file using the method
close().Now open the file
"output.txtx"in read-only mode and print to screen its content.use
open()to open the file"poetry.txt"in write-only memory. Next, write in the file the strings of the following list, one per line:verses = [ "S'i fosse fuoco, arderei 'l mondo" "s'i fosse vento, lo tempestarei" ]
Next, do the same opening twice the file
"poetry2.txt"in write-only mode (appending), and each time write one of the two verses.What does it happen when we re-open
"poetry2.txt"in mode"w"and we close it immediately?Write a module
trick.pythat prints to screen its code.Curiosity: we just wrote (cheating!) a Quine program.
Python: Input-Output (Solutions)¶
Raw input¶
Solution:
food = raw_input("What is your favourite food? ") print "I also like", food
Solution:
a_and_b = raw_input("Write two integers: ") words = a_and_b.split() a = int(words[0]) b = int(words[1]) answer = raw_input("how much is " + str(a) + " " + str(b) " ? ") result = int(answer) print a + b == answer
Solution:
key = raw_input("give me a key: ") value = raw_input("give me a value: ") dictionary = {key: value} # or dictionary = {} dictionary[key] = value print "dictionary =", dictionary
Solution:
name = raw_input("Give me your full name: ") fixed_words = [word[0].upper() + word[1:].lower() for word in name.split()] print "Your name is:", " ".join(fixed_words)
Filesystem¶
Solution:
f = open("data/aatable", "r") # or f = open("data/aatable") rows = f.readlines() print type(rows) # list print type(rows[0]) # str print len(rows) f.close()
Solution:
f = open("data/aatable") first_row = f.readline() print "The first row is: ", first_row remaining_rows = f.readlines() print "another", len(remaining_rows), " rows are left" remaining_rows_bis = f.readlines() print "then, another", len(remaining_rows_bis), "rows are left" # In the last case, 0 rows should be left: the first # readlines() already read all the lines of f f.close()
Solution:
f = open("output.txt", "w") f.write("check one two three check") f.close() g = open("output.txt", "r") print g.readlines() g.close()
Solution:
verses = [ "S'i fosse fuoco, arderei 'l mondo" "s'i fosse vento, lo tempestarei" ] f = open("poetry.txt", "w") f.write("\n".join(verses)) f.close()
Now let’s try with
"a":f2 = open("poetry2.txt", "a") f2.write(verses[0] + "\n") f2.close() f2 = open("poetry2.txt", "a") f2.write(verses[1] + "\n") f2.close()
And if we use
"w"on"poetry2.txtx":f = open("poetry2.txt", "w") # Here we do absolutely nothing to f, we just close it f.close()
we can see that now
"poetry2.txt"is empty! This happens as a consequence of using"w"instead of"a".Let’s write in the file
trick.py:myself = open("trick.py") print myself.read() myself.close()
Let’s execute the file to verify that it’s working as we want: from a shell, let’s write:
python trick.py
Python: Complex Statements¶
Conditional code: if¶
The if/elif/else statements allow to write code that gets
executed if and only if some condition is satisfied.
For instance:
if condition:
print "the condition is True"
executes the print statement if and only if condition evaluates to
True.
We can also have multiple mutually exclusive alternatives:
if condition:
print "the condition is True"
else:
print "the condition is not True"
Here only one branch is executed, based on the value of condition.
The same is true here:
if condition1:
print "condition1 is True"
elif condition2:
print "condition1 is not True"
print "but condition2 is!"
elif condition3:
print "condition1 and condition2 are not True:
print "but condition3 is!"
else:
print "no condition is True"
The if, elif and else form a “chain”: only one of the branches
is executed.
Example. Suppose we have two Booleans c1 and c2. Let’s see in
detail which lines are executed based on the value of the two variables:
# c1 c2 | c1 c2 | c1 c2 | c1 c2
# True True | True False | False True | False False
# ----------+------------+------------+------------
print "begin" # yes | yes | yes | yes
if c1: # yes | yes | yes | yes
print "1" # yes | yes | no | no
elif c2: # no | no | yes | yes
print "2" # no | no | yes | no
else: # no | no | no | yes
print "none" # no | no | no | yes
print "end " # yes | yes | yes | yes
Let’s break the above into pieces:
- if
c1isTrue, then the value ofc2does not matter: neither theelif c2nor theelseare executed. - if
c1isFalse,c2decides whether theelif c2is executed or theelseis.
In other words, "c1" and "c2" are evaluated sequentially.
Assume that, instead, we want to print "2" independently of whether c1
is True or not. The only way to do that is to avoid the elif‘s:
print "begin"
if c1:
print "1"
if c2:
print "2"
if not c1 and not c2:
print "0"
Here the if‘s do not form a chain anymore: they are independent of
one another!
Example. Python uses the indentation to decide which code is “inside”
the if and which code is “outside”.
Let’s write a short program to check whether the user is a mentalist:
print "I am thinking of a number between 1 and 10"
is_mentalist = int(raw_input("which one? ")) == 72
print "Computing..."
if is_mentalist:
print "CONGRATULATIONS!!!"
print "you are a mentalist"
else:
print "thanks for playing"
print "better luck next time"
print "done"
In this example, the print statements that are indented after the if
and the else are “inside”: they are conditional on is_mentalist.
The other print‘s are “outside”: they are executed unconditionally.
Example. This code opens a file and checks whether it is a “valid” FASTA
file. In order to do so, it checks whether (1) the file is empty, and (2) the
file contains lines that start with the ">" character:
lines = open("data/prot-fasta/1A3A.fasta").readlines()
if len(lines) == 0:
print "the file is empty"
else:
first_characters = [line[0] for line in lines]
if not ">" in first_characters:
print "not a fasta file"
else:
print "a fasta file"
print "done"
Quiz:
- Can the code print both that the file is empty and that the file is valid?
- Can the code not print
"done"?- If the file is actually empty, what is the value of the variable
first_charactersat the end of the execution?- Can I simplify the code by using an
elifstatement?
Iterative code: for¶
The for statement allows to write code that is executed multiple times.
In particular, the code inside the for is executed once for each and
every element in a collection (i.e. string, list, tuple, dictionary).
The abstract syntax is:
collection = range(10) # a list, for instance
for element in collection:
body(element)
This for iterates over all the elements element in the collection and
executes the body(element) block on each of them.
Just like for list comprehensions, the element variable is defined by the
for loop. At the Nth iteration, element will refer to the Nth element
of collection.
The flow of the execution can be modified with the break and continue
statements, see below for details.
Warning
If collection is a:
str, theforiterates over the characters.list, theforiterates over the elements.tuple, theforiterates over the elements.dict, theforiterates over the keys.
Example. This for:
l = [1, 25, 6, 27, 57, 12]
for number in l:
print number
iterates over all elements of l, from beginning to end. At each iteration,
the value of number changes (as shown by the print).
The above for is equivalent to this code:
number = l[0]
print number
number = l[1]
print number
number = l[2]
print number
# etc.
except that it is a lot shorter!
Example. Let’s compute the sum of all elements of the previous list.
We can do that by modifying the for as follows:
l = [1, 25, 6, 27, 57, 12]
s = 0
for number in l:
s = s + number # equiv. s += number
print "the sum is", s
Here s plays the role of a support variable. It is initialized to 0
just before the loop. Then, each number in l is added, in turn, to s.
By the end of the for,
The above code is equivalent to:
s = 0
number = l[0]
s += number
number = l[1]
s += number
# etc.
Example. Now let’s find the largest element in the list. The idea is:
- We use a support variable
largest_so_farthat always (at all iterations) holds the largest element found so far. It is initialized to some sensible value. - We use a
forto iterate over all elements of the list. - If the current element is smaller than or equal than
largest_so_far, the latter is left untouched. - Otherwise,
largest_so_faris updated to reference the current element.
Once the for is done, i.e. after iterating over the very last element
of the list, largest_so_far will hold the largest element in the list.
Let’s write:
l = [1, 25, 6, 27, 57, 12]
# l[0] is a sensible initial value
largest_so_far = l[0]
for number in l[1:]:
if number > largest_so_far:
largest_so_far = number
print "the maximum is", largest_so_far
Example. Given the following table (list of strings):
table = [
"protein domain start end",
"YNL275W PF00955 236 498",
"YHR065C SM00490 335 416",
"YKL053C-A PF05254 5 72",
"YOR349W PANTHER 353 414",
]
I want to convert it to a dictionary like this:
data = {
"YNL275W": ("PF00955", 236, 498),
"YHR065C": ("SM00490", 335, 416),
"YKL053C-A": ("PF05254", 5, 72),
"YOR349W": ("PANTHER", 353, 414)
}
The keys are taken from the first column, while the values are the remaining columns. Let’s write:
# the dictionary is initially empty
data = {}
# for each line in the table (except the header)
for line in table[1:]:
words = line.split()
protein = words[0]
domain = words[1]
pos0 = int(words[2])
pos1 = int(words[3])
# update the dictionary
data[protein] = (domain, pos0, pos1)
Example. The break statement allows to interrupt the for. For
instance:
path = raw_input("write a path to a file: ")
lines = open(path).readlines()
for line in lines:
line = line.strip()
print "processing:", line
# if the line is "STOP", we break out of the
# for loop: the remaining lines are not
# processed
if line == "STOP":
break
# <--- when Python encounters the break statement,
# it "jumps" here
This code reads a text file and prints each line on screen. However, as soon
as it finds a "STOP" line, it executes the break, which exits the
for loop.
All the lines coming after the "STOP" line are not processed.
Example. The continue statement allows to skip to the next iteration,
skipping the remainder of the code in this iteration. For instance:
path = raw_input("write a path to a file: ")
lines = open(path).readlines()
for line in lines:
line = line.strip()
print "processing:", line
if line == "CONTINUE":
continue
# <--- if the continue is executed, the code from here...
print "this is not a CONTINUE line"
# <--- ... to here is not executed
reads a user-provided text file. It prints every line in turn. If the line
is "CONTINUE", the continue statement skips over the second print.
The for cycle restarts from the next line.
Iterative code: while¶
The while statement allows to write code that repeats as long as a certain
condition is true. The while stops iterating as soon as the condition is
not true anymore.
The abstract syntax is:
while condition:
do_stuff()
condition = check_condition()
As with the for, the break and continue statements can be used to
modify the flow of the execution.
Note
The big difference between the for and while statements is:
for element in collection:executes N times, where N is the length ofcollection.while condition:executes an indefinite number of times, that is, as long as the condition is true.
Example. The while statement is useful when the value of condition
can not be known beforehand, for instance when interacting with a user.
Let’s write a while that asks the user whether she wants to stop, and
keeps asking as long as the user does not reply "yes":
while raw_input("do you want me to stop? ") != "yes":
print "Then I'll keep going!"
Example. Let’s see another simple example with a break:
while True: # this is an infinite while!
ans = raw_input("what is the capital of Italy? ")
if ans.lower() == "rome":
print "correct"
break
print "try again"
# <--- the break jumps here
print "done"
I can not really do the same with a for loop!
Let’s make the code ask the user whether she actually wants to retry:
while True:
ans = raw_input("what is the capital of Italy? ")
if ans.lower() == "rome":
print "correct"
break
ans = raw_input("try again? ")
if ans.lower() == "no":
print "allright"
break
Nested code¶
Now that we know what if, for and while do, we can combine them in
arbitrary ways by properly nesting (that is, indenting) the statements.
Example. Let’s write a simulator of a two-hand clock (hours and minutes):
for h in range(24):
for m in range(60):
print "time =", h, ":", m
Here the external for iterates over the 24 hours; for each hour, the inner
for iterates over the 60 minutes.
Every time the internal for completes, the external for completes
one iteration.
Let’s extend the simulator to a hour-minutes-seconds clock:
for h in range(24):
for m in range(60):
for s in range(60):
print "time =", h, ":", m, ":", s
Of course, it is possible to take days into consideration by adding one more
external loop that iterates over range(1, 366).
Example. I want to check whether a list contains repeated elements, and if it does, what are their positions. Starting from:
numbers = [5, 9, 4, 4, 9, 2]
we can use two nested for statements to iterate over the pairs of
elements of numbers.
For every element (let’s say the one in position i), I want to check
whether the following elements (those in position i+1 to len(numbers) - 1)
match.
A picture is worth a thousand words:
+---+---+---+---+---+---+
| 5 | 9 | 4 | 4 | 9 | 2 |
+---+---+---+---+---+---+
^
i
\__________________/
the possible positions of the 2nd element
+---+---+---+---+---+---+
| 5 | 9 | 4 | 4 | 9 | 2 |
+---+---+---+---+---+---+
^ ^
i MATCH!
\______________/
the possible positions of the 2nd element
+---+---+---+---+---+---+
| 5 | 9 | 4 | 4 | 9 | 2 |
+---+---+---+---+---+---+
^ ^
i MATCH!
\__________/
the possible positions of the 2nd element
Let’s write:
matches = []
for i in range(len(numbers)):
# the number at position i
number_at_i = numbers[i]
for j in range(i + 1, len(numbers)):
# the number at position j
number_at_j = numbers[j]
# do they match?
if number_at_i == number_at_j:
# they do! let's store their
# positions
matches.append((i, j))
print matches
Let’s verify whether matches actually identifies pairs of identical
elements:
for pair in matches:
number_at_i = numbers[pair[0]]
number_at_j = numbers[pair[1]]
print number_at_i == number_at_j
Example. Given the contents of a FASTA a file:
>>> lines = open("data/prot-fasta/3J01.fasta").readlines()
>>> print lines
[
">3J01:0|PDBID|CHAIN|SEQUENCE",
"AVQQNKPTRSKRGMRRSHDALTAVTSLSVDKTSGEKHLRHHITADGYYRGRKVIAK",
">3J01:1|PDBID|CHAIN|SEQUENCE",
"AKGIREKIKLVSSAGTGHFYTTTKNKRTKPEKLELKKFDPVVRQHVIYKEAKIK",
">3J01:2|PDBID|CHAIN|SEQUENCE",
"MKRTFQPSVLKRNRSHGFRARMATKNGRQVLARRRAKGRARLTVSK",
">3J01:3|PDBID|CHAIN|SEQUENCE",
# ...
]
I want to convert lines into a dictionary that maps from each header (key)
to the corresponding sequence (value). Let’s write:
sequence_of = {}
for line in lines:
# remove newlines and spaces around the line
line = line.strip()
if line.startswith(">"):
# this is a header, store it for later use
header = line
else:
# this is a sequence
sequence = line
# now let's use the header we read at the
# *previous* iteration and the sequence we
# got at the *current* iteration to update
# dictionary
sequence_of[header] = sequence
# we are done; print the dictionary
print sequence_of
This code works as long as the sequences only span one line. However, this is
not the case for the FASTA file we have. Looking closer, we see that lines
includes these lines:
lines = [
# ...
">3J01:5|PDBID|CHAIN|SEQUENCE",
"MAKLTKRMRVIREKVDATKQYDINEAIALLKELATAKFVESVDVAVNLGIDARKSDQNVRGATVLPHGTGRSVRVAVFTQ",
"GANAEAAKAAGAELVGMEDLADQIKKGEMNFDVVIASPDAMRVVGQLGQVLGPRGLMPNPKVGTVTPNVAEAVKNAKAGQ",
"VRYRNDKNGIIHTTIGKVDFDADKLKENLEALLVALKKAKPTQAKGVYIKKVSISTTMGAGVAVDQAGLSASVN",
# ...
]
So there is one header, then a multi-line sequence.
Unfortunately with the above code, the first line of the sequence is overwritten by the second line of the sequence, which is then overwritten by the third line of the sequence. In other words, only the last line of a multi-line sequence makes it to the dictionary.
Let’s fix the code:
sequence_of = {}
for line in lines:
line = line.strip()
if line.startswith(">"):
header = line
else:
sequence = line
# the first time we encounter a header, we
# associate it to an empty string
if not sequence_of.has_key(header):
sequence_of[header] = ""
# now we take whatever sequence is associated
# to the header and concatenate it with the
# current line
old_sequence = sequence_of[header]
new_sequence = old_sequence + sequence
sequence_of[header] = new_sequence
A shorter version:
for line in lines:
line = line.strip()
if line.startswith(">"):
header = line
else:
if not sequence_of.has_key(header):
sequence_of[header] = line
else:
sequence_of[header] += line
Example. Same setup as before. Some anonymous jester wrote the FASTA file wrong: sequences come before the corresponding headers. For instance:
wrong_fasta = [
# first sequence
"AVQQNKPTRSKRGMRRSHDALTAVTSLSVDKTSGEKHLRHHITADGYYRGRKVIAK",
# first header
">3J01:0|PDBID|CHAIN|SEQUENCE",
# second sequence
"AKGIREKIKLVSSAGTGHFYTTTKNKRTKPEKLELKKFDPVVRQHVIYKEAKIK",
# second header
">3J01:1|PDBID|CHAIN|SEQUENCE",
]
Our code of course relies on the header coming before the sequence – so it does not work in this case. How do make it work again?
We have to rewrite the code based on the fact that the header is not known when we get the sequence. It is true however that we know the sequence when we get the header.
Let’s write:
sequence_of = {}
# this variable is used to hold the multi-line
# sequence we have seen `so far'
# it is initialized with an empty string, because
# we have not seen any sequence yet!
latest_sequence_seen = ""
for line in lines:
line = line.strip()
if line.startswith(">"):
# this is a header line. at this point the
# sequence is known, and we can update the
# dictionary
sequence_of[line] = latest_sequence_seen
# reset the latest sequence seen (so not to
# mix the sequences of different proteins/genes)
latest_sequence_seen = ""
else:
# this is a sequence line. we do not know
# the header yet. let's just add this sequence
# to the
latest_sequence_seen += line
print sequence_of
Exercises: if¶
Warning
Do not forget the colon : after the if, else, etc.
Writing an if without the colon, e.g.:
>>> answer = raw_input("do you like yellow? ")
>>> if answer == "yes"
is an error. As soon as I enter the last line of code, Python gets upset:
File "<stdin>", line 1
if answer == "yes"
^
SyntaxError: invalid syntax
and refuses to execute the code.
Warning
Watch out for wrong indentation levels!
In Python, wrong indentation means wrong code.
The code may still “run”, but it may compute the wrong thing.
In some cases, it is easy to spot what’s wrong. For instance, here Python immediately raises an error:
>>> answer = raw_input("do you like yellow? ")
>>> if answer == "yes":
>>> print "you said:"
>>> print "yes"
File "<stdin>", line 4
print "yes"
^
IndentationError: unexpected indent
In other cases the error can be much more subtle and difficult to find. See below the section on nested statements.
Ask the user a number (with
raw_input()). If the number is even, print"even"; print"odd"otherwise.Hint.
raw_input()always returns a string.Ask the user a float. If the number is in the interval [-1, 1], print
"okay". Do not print anything otherwise.Hint. Are
elif/elsenecessary in this case?Ask the user two integers. If the first one is larger than the second one, print
"first". If the second is larger than the first, print"second". Otherwise, print"neither".Given the dictionary:
horoscope_of = { "January": "extreme luck", "February": "try to be born again", "March": "kissed by fortune", "April": "lucky luke", }
ask the user her birth month. If the month appears (as a key) in the dictionary, print the corresponding horoscope. Otherwise, print
"not available".Ask the user a path to an existing file, and read the contents using
readlines(). Then print:- If the file is empty, the string
"empty" - If the file has less than 100 lines,
"short", as well as the number of lines. - If the file has between 100 and 1000 lines,
"average"and the number of lines. - Otherwise, print
"large"and the number of lines.
The message must be printed on a single line.
- If the file is empty, the string
Using two calls to
raw_input(), ask the user two triples of floats. The two triples represent 3D coordinates: x, y, z.If all coordinates are non-negative, print the Euclidean distance between the two points. Do not print anything otherwise.
Hint: the Euclidean distance is given by \(\sqrt{(x_1 - x_2)^2 + (y_1 - y_2)^2 + (z_1 - z_2)^2}`\)
Read this code:
number = int(raw_input("write a number: ")) if number % 3 == 0: print "divisible by 3" elif number % 3 != 0: print "not divisible by 3" else: print "dunno"
Can it actually print
"dunno"?Read this code:
number = int(raw_input("write a number: ")) if number % 2 == 0: print "divisible by 2" if number % 3 == 0: print "divisible by 3" if number % 2 != 0 and number % 3 != 0: print "dunno"
Can it actually print
"dunno"?Ask the user whether he wants to perform a
"sum"or a"product".If the user asks for a
"sum", ask for two numbers, sum them, and print the result.Otherwise, if the user asks for a
"product", ask for two numbers, multiply them, and print the result.If the user replies neither
"sum"or"product", do nothing.
Exercises: for and while¶
Write a
forcycle to perform the following tasks:Print to screen the elements of
range(10), one for each row.Print to screen the square of the elements of
range(10), one for each row.Print to screen the sum of squares of
range(10).Print to screen the product of the elements of
range(1,10).Given the dictionary:
volume_of = { "A": 67.0, "C": 86.0, "D": 91.0, "E": 109.0, "F": 135.0, "G": 48.0, "H": 118.0, "I": 124.0, "K": 135.0, "L": 124.0, "M": 124.0, "N": 96.0, "P": 90.0, "Q": 114.0, "R": 148.0, "S": 73.0, "T": 93.0, "V": 105.0, "W": 163.0, "Y": 141.0, }
containing the volume of each amino acid, print to screen the sum of all the values.
Given the dictionary:
volume_of = { "A": 67.0, "C": 86.0, "D": 91.0, "E": 109.0, "F": 135.0, "G": 48.0, "H": 118.0, "I": 124.0, "K": 135.0, "L": 124.0, "M": 124.0, "N": 96.0, "P": 90.0, "Q": 114.0, "R": 148.0, "S": 73.0, "T": 93.0, "V": 105.0, "W": 163.0, "Y": 141.0, }
containing the volume of each amino acid, and the FASTA string:
fasta = """>1BA4:A|PDBID|CHAIN|SEQUENCE DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVV"""
print to screen the total volume of the amino acids of the protein sequence.
Hint. First, you should extract the amino acid sequence from
fasta, then, for each character of the sequence (for character in sequence) get from the dictionary the corresponding volume and add it to the total.Find the minimum value of the list
[1, 25, 6, 27, 57, 12].Hint. See the previous example about finding the maximum of a list, and adapt it to the new logic (auxiliary variable
minimum_so_far).Find both the maximum and the minimum of the list
[1, 25, 6, 27, 57, 12].Hint. You should create two auxiliary variables:
maximum_so_farandminimum_so_far.Given the nucleotide sequence:
sequence = "ATGGCGCCCGAACAGGGA"
compute the list of all its codons (0 offset for reading frame). The solution should be:
["ATG", "GCG", "CCC", "GAA", "CAG", "GGA"]
Hint: you should iterate on the result of
range(0, len(sequence), 3)and add at each step the sequence of a codon to a previously created empty list.Given the text (in FASTA format):
text = """>2HMI:A|PDBID|CHAIN|SEQUENCE PISPIETVPVKLKPGMDGPKVKQWPLTEEKIKALVEICTEMEKEGKISKI >2HMI:B|PDBID|CHAIN|SEQUENCE PISPIETVPVKLKPGMDGPKVKQWPLTEEKIKALVEICTEMEKEGKISKI >2HMI:C|PDBID|CHAIN|SEQUENCE DIQMTQTTSSLSASLGDRVTISCSASQDISSYLNWYQQKPEGTVKLLIYY >2HMI:D|PDBID|CHAIN|SEQUENCE QITLKESGPGIVQPSQPFRLTCTFSGFSLSTSGIGVTWIRQPSGKGLEWL >2HMI:E|PDBID|CHAIN|SEQUENCE ATGGCGCCCGAACAGGGAC >2HMI:F|PDBID|CHAIN|SEQUENCE GTCCCTGTTCGGGCGCCA"""
return the dictionary
sequence_of, having as keys the names of the sequences (the first will be2HMI:A, the second2HMI:B, and so on), and as values the corresponding sequences.The result should resemble this:
sequence_of = { "2HMI:A": "PISPIETVPVKLKPGMDGPKVKQW...", "2HMI:B": "PISPIETVPVKLKPGMDGPKVKQW...", # ... }
Hint. You should first split
textin lines. Next, you should iterate on lines: if the line is a header, you should save the name of the sequence; otherwise, you should update the dictionary with the name you got from the previous line, and the sequence you have in the current line.
Write a
whilecycle performing the following task:- keep asking the user to write
"STOP". If the user writes``”STOP”`` (in upper case) the cycle terminates, otherwise it prints"you must write 'STOP'..."and continues. - as before, but the cycle terminates also if the user writes
"stop"in lower case.
- keep asking the user to write
What is printed to screen when executing the following code?
for number in range(10): print "processing the element", number
for number in range(10): print "processing the element", number break
for number in range(10): print "processing the element", number continue
for number in range(10): print number if number % 2 == 0: break
for number in range(10): if number % 2 == 0: break print number
condition = False while condition: print "the condition is true"
condition = False while condition: print "the condition is true" condition = True
condition = True while condition: print "the condition is true"
numbers = range(10) i = 0 while i < len(numbers): print "position", i, "contains the element", numbers[i]
lines = [ "line 1", "line 2", "line 3", "", "line 5", "line 6", ] for line in lines: line = line.strip() if len(line) == 0: break else: print "I read:", line
Given the tuple:
numbers = (0, 1, 1, 0, 0, 0, 1, 1, 2, 1, 2)
write a cycle that iterates on
numbers, stopping whenever finding the value2and printing to screen its position.Given the tuple:
strings = ("000", "51", "51", "32", "57", "26")
write a cycle that iterates on
strings, stopping whenever finding a string containing the character"2", and printing to screen position and value of the string that stopped the cycle.The solution should be: position
4, value"32".Adapted from example 2.4 in [book2] : create a random nucleotide sequence, with length defined by the user, and print it to screen.
Hint. The
randommodule allows you to create random numbers. It provides a number of tools to manage random objects. For example, therandint(i,j)function generates a number betweeniandjwith equal probabilities, for example:import random index = random.randint(0,3) #index can be used to select the nucleotide at each position
Exercises: nested statements¶
Given the matrix:
n = 5 matrix = [range(n) for i in range(n)]
write a double
forcycle printing to screen all the elements of matrix, one for each line.Given the matrix:
n = 5 matrix = [range(n) for i in range(n)]
what do the following fragments of code print?
for row in matrix: for element in row: print element
sum = 0 for row in matrix: for element in row: sum = sum + element print sum
for i in range(len(matrix)): row = matrix[i] for j in range(len(row)): element = row[j] print element
for i in range(len(matrix)): for j in range(len(matrix[i])): print matrix[i][j]
dunno = [] for i in range(len(matrix)): for j in range(len(matrix[i])): if i == j: dunno.append(matrix[i][j]) print " ".join([str(x) for x in dunno])
Given the list:
numbers = [8, 3, 2, 9, 7, 1, 8]
write a double
forcycle printing to screen all the pairs of elements ofnumbers.Modify the solution of the last exercise so that, if the pair
(i,j)has been already printed, then the symmetric pair(j,i)is not printed.Hint. See the example above.
Do the same as in the last exercise with the following list:
strings = ["I", "am", "a", "list"]
Given the list:
numbers = range(10)
write a double
forprinting to screen only pairs of elements ofnumberswhere the second element of the pair is twice the first.The result will be:
0 0 1 2 2 4 ...
Given the list:
numbers = [8, 3, 2, 9, 7, 1, 8]
write a double
forcycle iterating on all element pairs ofnumbersand printing to screen the pairs whose sum is10.(Printing “repetitions” such as
8 + 2and2 + 8is allowed.)The result will be:
8 2 3 7 2 8 9 1
Hint. There is an example showing how to iterate on all pairs of elements of a list. It is sufficient to modify this example.
As before, but instead of printing to screen, store the pairs of elements whose sum is
10in a listlist_of_pairs.The result will be:
>>> list_of_pairs [(8, 2), (3, 7), (2, 8), 9, 1)]
Given the lists:
number_1 = [5, 9, 4, 4, 9, 2] number_2 = [7, 9, 6, 2]
write a double
forcycle iterating on the two lists and printing to screen values and positions of all the elements ofnumber_1appearing also innumber_2.The result will be:
positions: 1, 1; repeated value: 9 positions: 4, 1; repeated value: 9 positions: 5, 3; repeated value: 2
As before, but instead of printing to screen, store positions and value in a list of triplets like this:
(position_1, position_2, repeated_values).Given the matrix:
n = 5 matrix = [range(n) for i in range(n)]
write a double
forcycle finding the higher element.Hint. It is sufficient to adapt the code that finds the maximum-minimum of a list (with one dimension) to a matrix (with two dimensions).
Given the list of nucleotide sequences:
sequences = [ "ATGGCGCCCGAACAGGGA", "GTCCCTGTTCGGGCGCCA", ]
we want to obtain a list containing, for each sequence in
sequences, the list of its triplets.Hint. You can re-use a previous exercise.
Given the list:
numbers = [5, 9, 4, 4, 9, 2]
write a code that counts the number of occurrences of each element and store the result in a dictionary, similar to this:
num_occurrences = { 5: 1, 9: 2, 4: 2, 2: 1, }
Hint. You can modify one of the previous examples so that, instead of saving the position of occurrences, increases the number of occurrences in
num_occurrences.Hint. Note that if the key
5is not in the dictionary, we cannot executenum_occurrences[5] += 1, sincenum_occurrences[5]doesn’t exist. See the example about reading a FASTA file.Given a list of gene clusters (lists), for example:
groups = [["gene1", "gene2"], ["gene3"], [], ["gene4", "gene5"]]
write a single cycle finding the biggest group and storing it in a variable
biggest_group_so_far.Hint: this task is similar to finding the minimum/maximum in a list of integers, but the auxiliary variable should contain the longer list found so far.
Given the list of sequences:
sequences_2HMI = { "A": "PISPIETVPVKLKPGMDGPKVKQWPLTEEKI", "B": "PISPIETVPVKLKPGMDGPKVKQWPLTEEKI", "C": "DIQMTQTTSSLSASLGDRVTISCSASQDISS", "D": "QITLKESGPGIVQPSQPFRLTCTFSGFSLST", "E": "ATGGCGCCCGAACAGGGAC", "F": "GTCCCTGTTCGGGCGCCA", }
write a
forcycle that (iterating on all the key-value pairs of the dictionary) returns a dictionary of histograms (that is dictionaries mapping amino acids to their number of occurrences) of each element ofsequences_2HMI.Hint. Calculating a histogram also requires a
forcycle: therefore you should expect to code two nestedforcycles.The result (a dictionary of dictionaries) should be like this:
histograms = { "A": { "P": 6, "I": 3, "S": 1, #... }, "B": { "P": 6, "I": 3, "S": 1, #... }, #... "F": { "A": 1, "C": 7, "G": 6, "T": 4, } }
Given the list of strings:
table = [ "protein domain start end", "YNL275W PF00955 236 498", "YHR065C SM00490 335 416", "YKL053C-A PF05254 5 72", "YOR349W PANTHER 353 414", ]
write a code that takes column names from the first row of
tableand:for each row creates a dictionary like this:
dictionary = { "protein": "YNL275W", "domain": "PF00955", "start": "236", "end":, "498" }
append the dictionary to a list.
Given:
alphabet_lo = "abcdefghijklmnopqrstuvwxyz" alphabet_up = alphabet_lo.upper()
write a cycle (
fororwhile) that, starting from an empty dictionary, insert all the key-value pairs:"a": "A", "b": "B", ...
in other words, the dictionary maps from the i-th character of
alphabet_minto the i-th character ofalphabet_max.Next, use the dictionary to implement a
forcycle that, given an arbitrary string, for example:string = "I am a string"
returns the same result of
string.upper().Write a module that asks the user for the path to two text files, and print to screen the rows of the two files, one by one, next to each other: the rows of the first file should be printed on the left, the rows of the second to the right.
If the first file contains:
first row second row
and the second:
ACTG GCTA
the result will be:
first row ACTG second row GCTA
Hint. Note that the two files could be of different length. In that case (optionally) missing lines should be printed as if they were empty lines.
Write a module that, given the file
data/dna-fasta/fasta.1:- Read the contents of the FASTA file in a dictionary.
- Calculate how many times each nucleotide appears in each sequence.
- Calculate the GC-content of each sequence.
- Calculate the AT/GC-ratio of each sequence.
Given the genetic code, provided as a dictionary, write a program that reads an RNA sequence from a FASTA file as a string, translates the sequence in each possible reading frame and print it to screen:
codon_table = { 'GCU':'A','GCC':'A','GCA':'A','GCG':'A','CGU':'R', 'CGC':'R','CGA':'R','CGG':'R','AGA':'R','AGG':'R', 'UCU':'S','UCC':'S','UCA':'S','UCG':'S','AGU':'S', 'AGC':'S','AUU':'I','AUC':'I','AUA':'I','AUU':'I', 'AUC':'I','AUA':'I','UUA':'L','UUG':'L','CUU':'L', 'CUC':'L','CUA':'L','CUG':'L','GGU':'G','GGC':'G', 'GGA':'G','GGG':'G','GUU':'V','GUC':'V','GUA':'V', 'GUG':'V','ACU':'T','ACC':'T','ACA':'T','ACG':'T', 'CCU':'P','CCC':'P','CCA':'P','CCG':'P','AAU':'N', 'AAC':'N','GAU':'D','GAC':'D','UGU':'C','UGC':'C', 'CAA':'Q','CAG':'Q','GAA':'E','GAG':'E','CAU':'H', 'CAC':'H','AAA':'K','AAG':'K','UUU':'F','UUC':'F', 'UAU':'Y','UAC':'Y','AUG':'M','UGG':'W', 'UAG':'*','UGA':'*','UAA':'*' }
Exercise 5.4 in in [book2]. Write a sequence-based predictor for protein secondary structure elements. Use the following dictionaries of preferences for alpha helices and beta sheets:
helix_propensity = { 'A':1.450, 'C':0.770, 'D':0.980, 'E':1.530, 'F':1.120, 'G':0.530, 'H':1.240, 'I':1.000, 'K':1.070, 'L':1.340, 'M':1.200, 'N':0.730, 'P':0.590, 'Q':1.170, 'R':0.790, 'S':0.790, 'T':0.820, 'V':1.140, 'W':1.140, 'Y':0.610 } sheet_propensity = { 'A':0.970, 'C':1.300, 'D':0.800, 'E':0.260, 'F':1.280, 'G':0.810, 'H':0.710, 'I':1.600, 'K':0.740, 'L':1.220, 'M':1.670, 'N':0.650, 'P':0.620, 'Q':1.230, 'R':0.900, 'S':0.720, 'T':1.200, 'V':1.650, 'W':1.190, 'Y':1.290 }
Hint. Scan the input sequence residue by residue and replace each residue with
H (helix) if its helix_propensity ≥ 1 and its helix_propensity > sheet_propensity, with S (sheet) if
its sheet_propensity ≥ 1 and its helix_propensity < sheet_propensity, and with L (loop) otherwise.
Read the input sequence from a FASTA file, and print (or write to a file) the input and output sequences, one on top of the
other.
Python: complex statements (Solutions)¶
Conditional code: if¶
Solution:
number = int(raw_input("write a number: ")) if number % 2 == 0: print "even" else: print "odd"
We use
else, since even and odd are the only two possibilities.A way to make a third option explicit would be:
if number % 2 == 0: print "even" elif number % 2 == 1: print "odd" else: print "impossible!"
but the code in
elsewill never be executed for any value ofnumber!Since the two options are mutually exclusive, we can also write:
if number % 2 == 0: print "even" if numero % 2 == 1: print "odd"
even without the``else``, one and only one of the
ifcan be executed.Solution:
number = float(raw_input("write rational: ")) if number >= -1 and number <= 1: print "okay"
we don’t need neither``elif`` (there is only one condition) neither
else(if the condition is false, we don’t need to do anything).Solution:
answer = raw_input("write two numbers separated by a space: ") words = answer.split() num1 = int(words[0]) num2 = int(words[1]) if num1 > num2: print "first" elif num2 > num1: print "second" else: print "neither"
Alternatively:
answer = raw_input("write two numbers separated by a space: ") numbers = [int(word) for word in answer.split()] if numbers[0] > numbers[1]: print "first" elif numbers[0] < numbers[1]: print "second" else: print "neither"
Solution:
horoscope_of = { "January": "extreme luck", "February": "try to be born again", "March": "kissed by fortune", "April": "lucky luke", } month = raw_input("tell me your birth month: ") if horoscope_of.has_key(month): print horoscope_of[month] else: print "not available"
Solution:
path = raw_input("write your path: ") lines = open(path, "r").readlines() if len(lines) == 0: print "empty" elif len(lines) < 100: print "short", len(lines) elif len(lines) < 1000: print "average", len(lines) else: print "large", len(lines)
Note that it’s not necessary to specify entirely the conditions: in the code we can shorten
100 < len(lines) < 1000withlen(lines) < 1000. We can do that, since when``len(lines)`` is lower than100the firstelifis executed: the secondelifis not even considered.Solution:
point1 = [float(word) for word in raw_input("write three coordinates: ").split()] point2 = [float(word) for word in raw_input("write three coordinates: ").split()] if point1[0] >= 0 and point1[1] >= 0 and point1[2] >= 0 and \ point2[0] >= 0 and point2[1] >= 0 and point2[2] >= 0: diff_x = point1[0] - point2[0] diff_y = point1[1] - point2[1] diff_z = point1[2] - point2[2] print "the distance is", (diff_x**2 + diff_y**2 + diff_z**2)**0.5
Note that
printis inside theif.Solution: we know that
numberis an arbitrary integer, chosen by the user:if number % 3 == 0: print "divisible by 3" elif numero % 3 != 0: print "not divisible by 3" else: print "dunno"
if,elifandelseform a chain: only one among them is executed.ifis executed if and only ifnumberis divisibile by three.elifis executed if and only if the previousifis not executed and ifnumberis not divisible by three.elseis execute whenever neitherifandelifare executed.
Since all numbers are either divisible by
3either not, there is no other possibility,elsewill never be executed.Therefore, the answer is no.
Solution: as before,
numberis an arbitrary integer. The code is:number = int(raw_input("write a number: ")) if number % 2 == 0: print "divisible by 2" if number % 3 == 0: print "divisible by 2" if number % 2 != 0 and number % 3 != 0: print "dunno"
Here we don’t have “chains” of
if,elifedelse: we have three independentif.- The first
ifis executed if and only ifnumberis divisible by two. - The second
ifis executed if and only ifnumberis divisible by three. - The third
ifis executed if and only ifnumberis not divisible by neither two and three.
If
numberis 6, divisible by both two and three, the first twoifwill be both executed, while the third won’t be.If
numberis 5, not divisible by neither two and three, the first twoifwill not be executed, but the third will be.Therefore, the answer is yes.
(There is no possibility to not execute neither of the three
if.)- The first
Solution:
answer = raw_input("sum or product?: ") if answer == "sum": num1 = int(raw_input("number 1: ")) num2 = int(raw_input("number 2: ")) print "the sum is", num1 + num2 elif answer == "product": num1 = int(raw_input("num1: ")) num2 = int(raw_input("num2: ")) print "the product is", num1 * num2
Using
iforelifwon’t change the execution of the program.We can simplify like this:
answer = raw_input("sum or product?: ") num1 = int(raw_input("number 1: ")) num2 = int(raw_input("number 2: ")) if answer == "sum": print "the sum is", num1 + num2 elif answer == "product": print "the product is", num1 * num2
Iterative code: for and while¶
Solutions:
Solution:
for number in range(10): print number
Solution:
for number in range(10): print number**2
Solution:
sum_of_squares = 0 for number in range(10): sum_of_squares = sum_of_squares + number**2 print sum_of_squares
Solution:
product = 1 # note that for the product the initial value should be 1! for number in range(1,10): product = product * number print product
Solution:
volume_of = { "A": 67.0, "C": 86.0, "D": 91.0, "E": 109.0, "F": 135.0, "G": 48.0, "H": 118.0, "I": 124.0, "K": 135.0, "L": 124.0, "M": 124.0, "N": 96.0, "P": 90.0, "Q": 114.0, "R": 148.0, "S": 73.0, "T": 93.0, "V": 105.0, "W": 163.0, "Y": 141.0, } sum_of_volumes = 0 for volume in volume_of.values(): sum_of_volumes = sum_of_volumes + volume print sum_of_volumes
Solution:
volume_of = { "A": 67.0, "C": 86.0, "D": 91.0, "E": 109.0, "F": 135.0, "G": 48.0, "H": 118.0, "I": 124.0, "K": 135.0, "L": 124.0, "M": 124.0, "N": 96.0, "P": 90.0, "Q": 114.0, "R": 148.0, "S": 73.0, "T": 93.0, "V": 105.0, "W": 163.0, "Y": 141.0, } fasta = """>1BA4:A|PDBID|CHAIN|SEQUENCE DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVV""" # Let's extract the sequence sequence = fasta.split("\n")[1] sum_of_volumes = 0 # for each character in the sequence ... for aa in sequence: volume_of_aa = volume_of[aa] sum_of_volumes = sum_of_volumes + volume_of_aa print sum_of_volumes
Solution: let’s adapt the code from the previous example:
list = [1, 25, 6, 27, 57, 12] minimum_so_far = list[0] for number in list[1:]: if number < minimum_so_far: minimum_so_far = number print "the minimum value is:", minimum_so_far
Solution: let’s combine the example and the previous exercise:
list = [1, 25, 6, 27, 57, 12] max = list[0] min = list[0] for number in list[1:]: if number > max: max = number if number < min: min = number print "minimum =", min, "maximum =", max
Solution:
range(0, len(sequence), 3)returns[0, 3, 6, 9, ...], containing the positions of the first character of all the triplets.Let’s write:
sequence = "ATGGCGCCCGAACAGGGA" # let's start from an empty list triplets = [] for pos_start in range(0, len(sequence), 3): triplets = sequence[pos_start:pos_start+3] triplets.append(triplets) print triplets
Solution:
text = """>2HMI:A|PDBID|CHAIN|SEQUENCE PISPIETVPVKLKPGMDGPKVKQWPLTEEKIKALVEICTEMEKEGKISKI >2HMI:B|PDBID|CHAIN|SEQUENCE PISPIETVPVKLKPGMDGPKVKQWPLTEEKIKALVEICTEMEKEGKISKI >2HMI:C|PDBID|CHAIN|SEQUENCE DIQMTQTTSSLSASLGDRVTISCSASQDISSYLNWYQQKPEGTVKLLIYY >2HMI:D|PDBID|CHAIN|SEQUENCE QITLKESGPGIVQPSQPFRLTCTFSGFSLSTSGIGVTWIRQPSGKGLEWL >2HMI:E|PDBID|CHAIN|SEQUENCE ATGGCGCCCGAACAGGGAC >2HMI:F|PDBID|CHAIN|SEQUENCE GTCCCTGTTCGGGCGCCA""" # first, let's split the text il lines lines = text.split("\n") # then, let's create an empty dictionary sequence_of = {} # now we can iterate on lines for line in lines: if line[0] == ">": # if the line is a header, we extract the sequence name name = line.split("|")[0] else: # the line contains the sequence, that we add to the dictionary, using the name extracted before as key sequence_of[name] = line print sequence_of
Solutions:
Solution:
while raw_input("write 'STOP': ") != "STOP": print "you must write 'STOP'..."
Solution:
while raw_input("write stop: ").lower() != "stop": print "you must write 'stop'..."
Solutions:
- Solution: all numbers in
range(10). - Solution: the number
0.breakimmediately interrupts theforcycle. - Solution: all numbers in
range(10).continuejumps to the next iteration, as Python automatically does when the instructions in theforcycle are finished. Sincecontinuein this case is right at the end of theforcycle, it doesn’t have any effect. - Solution: the number
0. In the first iteration, whennumberhas value0, first Python executesprint number, printing0; thenifis executed, and also thebreakinside theif, immediately interrupting theforcycle. - Solution: nothing. In the first iteration, when
numberhas value0,ifis executed and also thebreakinside theif, immediately interrupting theforcycle. Therefore,printis never executed. - Solution: nothing. Instructions inside the
whileare never executed, since the condition isFalse! - Solution: nothing. Instructions inside the
whileare never executed, since the condition isFalse! As a consequence, the linecondition = Trueis never executed. - Solution:
"the condition is true"an infinite number of times. Since the condition is alwaysTrue, thewhilenever stops iterating! - Solution: ten strings of the form
"position 0 contains the element 0","position 1 contains the element 1", and so on - Solution: all the elements of
lines(processed bystrip()) occurring before the first empty line:"line 1","line 2"and"line 3". As soon aslinehas value""(the fourth element oflines) theifis executed, andbreakinterrupts the cycle. Note that the fourth row is not printed.
- Solution: all numbers in
Solution:
numbers = (0, 1, 1, 0, 0, 0, 1, 1, 2, 1, 2) for i in range(len(numbers)): number_in_pos_i = numbers[i] if number_in_pos_i == 2: print "the position is", i break
Solution:
strings = ("000", "51", "51", "32", "57", "26") for i in range(len(strings)): string_in_pos_i = strings[i] if "2" in string_in_pos_i: print "position =", i, "value =", string_in_pos_i break
Solution:
length = int(raw_input("write the length of the sequence: ")) import random alphabet = "AGCT" sequence = "" for i in range(length): index = random.randint(0, 3) sequence = sequence + alphabet[index] print sequence
Nested code¶
Solution:
n = 5 matrix = [range(n) for i in range(n)] for line in matrix: for element in line: print element
Solution:
- All the elements of the matrix.
- The sum of all the elements of the matrix.
- Again, all the elements of the matrix.
- Again, all the elements of the matrix.
- The list of the elements on the diagonal.
Solution:
numbers = [8, 3, 2, 9, 7, 1, 8] for num_1 in numbers: for num_2 in numbers: print num_1, num_2
This code is very similar to the clock example!
Solution:
numbers = [8, 3, 2, 9, 7, 1, 8] already_printed_pairs = [] for i in range(len(numbers)): for j in range(len(numbers)): pair = (numbers[i], numbers[j]) # check wheter we already printed the symmetric pair if (pair[1], pair[0]) in already_printed_pairs: continue # this code will be executed if the pair has not been printed: # print the pair and update already_printed_pairs print pair already_printed_pairs.append(pair)
The solution is the same of the previous exercise.
Solution:
numbers = range(10) for element_1 in numbers: for element_2 in numbers: if 2 * element_1 == element_2: print element_1, element_2
Solution:
numbers = [8, 3, 2, 9, 7, 1, 8] for element_1 in numbers: for element_2 in numbers: if element_1 + element_2 == 10: print element_1, element_2
Solution:
numbers = [8, 3, 2, 9, 7, 1, 8] # first, let's create an empty list list_of_pairs = [] for element_1 in numbers: for element_2 in numbers: if element_1 + element_2 == 10: # update the list with append() list_of_pairs.append((element_1, element_2)) # finally, let's print the list print list_of_pairs
Solution:
numbers_1 = [5, 9, 4, 4, 9, 2] numbers_2 = [7, 9, 6, 2] # iteration on the *first* list for i in range(len(numbers_1)): num_in_pos_i = numbers_1[i] # iteration on the *second* list for j in range(len(numbers_2)): num_in_pos_j = numbers_2[j] if num_in_pos_i == num_in_pos_j: print "positions:", i, j, "; repeated value:", num_in_pos_i
Solution:
numbers_1 = [5, 9, 4, 4, 9, 2] numbers_2 = [7, 9, 6, 2] # first, let's create an empty list list_of_triplets = [] # iteration on the *first* list for i in range(len(numbers_1)): num_in_pos_i = numbers_1[i] # iteration on the *second* list for j in range(len(numbers_2)): num_in_pos_j = numbers_2[j] if num_in_pos_i == num_in_pos_j: # instead of printing, we update the list llist_of_triplets.append((i, j, num_in_pos_i)) # finally, let's print the list print list_of_triplets
Solution:
n = 5 matrix = [range(n) for i in range(n)] # let's initialize with the first element (any other element would be fine as well) max_element_so_far = matrix[0][0] # iteration... for line in matrix: for element in line: # we update max_element_so_far when we find a higher element, if element > max_element_so_far: max_element_so_far = element print max_element_so_far
Solution:
sequences = [ "ATGGCGCCCGAACAGGGA", "GTCCCTGTTCGGGCGCCA", ] # first, let's create an empty list result = [] # iteration for sequence in sequences: # split the current sequence in triplets triplets = [] for i in range(0, len(sequence), 3): triplets.append(sequence[i:i+3]) # append (*not* extend()!!!) the obtained triplets # to the list result result.append(triplets) # finally, let's print the list print result
Solution:
numbers = [5, 9, 4, 4, 9, 2] num_occurrences = {} for number in numbers: if not num_occurrences.has_key(number): num_occurrences[number] = 1 else: num_occurrences[number] += 1
alternatively:
numbers = [5, 9, 4, 4, 9, 2] num_occurrences = {} for number in numbers: if not num_occurrences.has_key(number): num_occurrences[number] = 0 num_occurrences[number] += 1
or, using
count():numbers = [5, 9, 4, 4, 9, 2] num_occurrences = {} for number in numbers: if not num_occurrences.has_key(number): num_occurrences[number] = numbers.count(number)
Note that in the last variant, the
ifline is optional (but not the following “content”!)Solution:
groups = [["gene1", "gene2"], ["gene3"], [], ["gene4", "gene5"]] # let's initialize with the first group biggest_group_so_far = groups[0] # iteration for grup in groups[1:]: if len(gropu) > len(biggest_group_so_far): biggest_group_so_far = group print biggest_group_so_far
Solution:
sequences_2HMI = { "A": "PISPIETVPVKLKPGMDGPKVKQWPLTEEKI", "B": "PISPIETVPVKLKPGMDGPKVKQWPLTEEKI", "C": "DIQMTQTTSSLSASLGDRVTISCSASQDISS", "D": "QITLKESGPGIVQPSQPFRLTCTFSGFSLST", "E": "ATGGCGCCCGAACAGGGAC", "F": "GTCCCTGTTCGGGCGCCA", } # let's start with an empty dictionary histograms = {} for key, sequence in sequences_2HMI.items(): # let's associate this key to an empty dictionary histograms[key] = {} for residue in sequence: if not histograms[key].has_key(residue): histograms[key][residue] = 1 else: histograms[key][residue] += 1 # let's print the result print histograms # let's print the result more clearly for key, histogram in histograms.items(): print key print histogram print ""
Solution:
table = [ "protein domain start end", "YNL275W PF00955 236 498", "YHR065C SM00490 335 416", "YKL053C-A PF05254 5 72", "YOR349W PANTHER 353 414", ] # as before, first let's extract column names from the first row column_names = table[0].split() # let's start from an empty list lines_as_dictionaries = [] # now, let's iterate on the other rows for line in table[1:]: # let's compile the dictionary for this row dictionary = {} words = line.split() for i in range(len(words)): # extract the corresponding word word= words[i] # extract the corresponding column name column_name = column_names[i] # update the dictionary dictionary[column_name] = word # having compiled the dictionary for this line, # we can update the list lines_as_dictionaries.append(dictionary) # finished! now let's print the result (one row at a time, # to make it easier to read) for row in lines_as_dictionaries: print row
Solution:
alphabel_lo = "abcdefghijklmnopqrstuvwxyz" alphabet_up = alfabeto_min.upper() # let's build the dictionary lo_to_up = {} for i in range(len(alphabel_lo)): lo_to_up[alphabel_lo[i]] = alphabel_up[i] string = "I am a string" # let's convert the string converted_chars = [] for character in string: if lo_to_up.has_key(character): # convert the alphabetic character converted_chars.append(lo_to_up[character]) else: # we don't convert it (e.g., it's not an alphabetic character) converted_chars.append(character) converted_string = "".join(converted_chars) print converted_string
Solution:
lines_1 = open(raw_input("path 1: ")).readlines() lines_2 = open(raw_input("path 2: ")).readlines() # we have to be careful, since the two files could be of different lengths! max_lines = len(lines_1) if len(lines_2) > max_lines: max_lines = len(lines_2) # iteration on the lines of both files for i in range(max_lines): # take the i-th line of the first file, if existent, if i < len(lines_1): line_1 = lines_1[i].strip() else: line_1 = "" # take the i-th line of the second file, if existent, if i < len(lines_2): line_2 = lines_2[i].strip() else: line_2 = "" print line_1 + " " + line_2
Solution:
# let's read the fasta file fasta_as_dictionary = {} for line in open("data/dna-fasta/fasta.1").readlines(): # let's clean the sequence line = line.strip() if line[0] == ">": header = line fasta_as_dictionary[header] = "" else: fasta_as_dictionary[header] += line # let's iterate on header-sequence pairs for header, sequence in fasta_as_dictionary.items(): print "processind", header # let's count the number of occurrences of each nucleotide count = {} for nucleotide in ("A", "C", "G", "T"): count[nucleotide] = sequence.count(nucleotide) print "nucleotide occurrences:", count # calculate gc-content gc_content = (count["G"] + count["C"]) / float(len(sequence)) print "GC content:", gc_content # calculate the AT/GC-ratio sum_at = count["A"] + count["T"] sum_cg = count["C"] + count["G"] at_gc_ratio = float(sum_at) / float(sum_cg) print "AT/GC-ratio:", at_gc_ratio
Python: Functions¶
Functions are named blocks of code. They take inputs and produce outputs.
The abstract syntax is:
def function(arg1, arg2, ...):
# the code
return result
Once a function is defined (as above), it can be called as follows:
the_result = function(value1, value2, ...)
The arguments (arg1, arg2, etc.) are variables that specify how many
inputs the function takes. The variable result is output from the function
to its caller.
Warning
The name of the variables I pass to the function has nothing to do with the name of the arguments.
In the code above, the values of the variables
valueXare visible from the inside the function asargX:arg1takes the value ofvalue1arg2takes the value ofvalue2- etc.
The name of the variable I use to store the result of the function has nothing to do with the name of the variable used inside the function to store the result.
In the code above, the value of the result is stored in the
resultvariable inside the function, but is stored inside thethe_resultvariable by the caller.the_resulttakes the value ofresult
Example:
def function(a, b):
r = a + b
return r
x = 5
y = 10
z = function(x, y)
print z
Here a takes its value from x, b from y; return r
makes the function return the value of r, which is assigned by the
caller to the variable z.
Example. Let’s define a function that takes two numbers and returns their sum:
def add(n, m):
return n + m
It can be used as follows:
result = add(4, 6)
print result # 10
result = add(6, 4)
print result # 10
By replacing the calls to add() with the code in the definition of the
add function, and substituting the arguments with the input values, we get
the equivalent code:
n = 4
m = 6
result = n + m
print result # 10
n = 6
m = 4
result = n + m
print result # 10
As with all Python-provided functions, I can also skip assigning the return
value to a variable (result in the code above):
print add(4, 6) # 10
Example. Let’s write a function print_sum() that prints the sum of
two numbers:
def print_sum(n, m):
print n + m
It can be used as follows:
print_sum(4, 6) # prints 10
print_sum(6, 4) # prints 10
Warning
Notice that there is no return statement in print_sum().
When return is omitted, the function automatically returns None:
result = print_sum(4, 6) # prints 10
print result # prints None
Warning
A function does nothing until it is called.
Consider the Python module test.py:
print "beginning"
def function():
print "I do stuff"
print "end"
Running it with the Python interpreter:
$ python test.py
produces the following output:
beginning
end
As you can see, the interpreter executes the code line by line. However,
while the function function() is defined in the middle of the module,
it is not called anywhere. Therefore it is not executed at all.
In order to actually call the function, let’s write:
print "beginning"
def function():
print "I do stuff"
function()
print "end"
This code prints:
beginning
I do stuff
end
as expected.
Example. Let’s write a function factorial() that computes the factorial
of an integer n:
Now, let’s compute the factorial of n normally (i.e. without defining
a new function):
fact = 1
for k in range(1, n + 1):
fact = fact * k
Now that we have the code for computing the factorial, it is easy to write a function that computes the factorial: it is sufficient to plug the above code inside a new function, as follows:
def factorial(n):
fact = 1
for k in range(1, n + 1):
fact = fact * k
return fact
And let’s check if it works:
print factorial(1) # 1
print factorial(2) # 2
print factorial(3) # 6
print factorial(4) # 24
print factorial(5) # 120
print factorial(6) # 720
Of course, the new function can be used like any of the Python-defined functions, e.g. in list comprehensions:
factorials = [factorial(n) for n in range(10)]
Warning
The name of the function, as well as the name of the arguments, are arbitrary: pick whichever name you find more fitting.
Quiz. What is the difference between this code:
def arith(op, a, b):
if op == "+":
return a + b
elif op == "*":
return a * b
else:
return None
print arith("+", 10, 10)
print arith("*", 2, 2)
and this code?:
def f(what, x, y):
if what == "+":
return x + y
elif what == "*":
return x * y
else:
return 0
print f("+", 10, 10)
print f("*", 2, 2)
Note
A function can return more than one result, as follows:
def multiresult():
result_1 = "first result"
result_2 = 0.12
result_3 = "something else"
return result_1, result_2, result_3
Internally, Python interprets the return statement as returning
a tuple. In practice, the above code is equivalent to:
def multiresult():
return ("first result", 0.12, "something else")
When I call a “multi-result” function, I can either put the resulting tuple into a variable and extract the various elements individually:
result = multiresult()
res1 = result[0]
print res1
res2 = result[1]
print res2
res3 = result[2]
print res3
or I can use the “automatic unpacking” feature of Python, as follows:
res1, res2, res3 = multiresult()
print res1
print res2
print res3
Warning
Variables have a scope, and in particular:
Variables declared outside the function are not visible the inside. [1]
If you want to pass one or more values from the outside to the function, pass them through the arguments.
Variables declared inside the function are not visible from the outside.
If you want to pass one or more values from the function to the external world, use the
returnstatement.
[1] There are exceptions to this rule; we will ignore them in this presentation.
Exampe. Consider this code:
def find_physical(triples):
"""Takes a mixed interaction protein network, example:
[("1A3A", "physical", "5ARM"),
("5JTD", "genetic", "5TGD")]
and extracts physical interacting protein pairs, example:
[("1A3A", "5ARM")]
"""
phys_pairs = []
for p1, relation, p2 in triples:
if relation == "physical":
phys_pairs.append((p1, p2))
# XXX I forgot to return `phys_pairs` here!
network = [
("1A3A", "physical", "5ARM"),
("5JTD", "genetic", "5TGD")
]
find_physical(network)
print phys_pairs
Here phys_pairs is declared inside the function: it is not visible
from the outside!
In order to fix this issue, I have to explicitly return it:
def find_physical(triples):
phys_pairs = []
for p1, relation, p2 in triples:
if relation == "physical":
phys_pairs.append((p1, p2))
return phys_pairs
network = [
("1A3A", "physical", "5ARM"),
("5JTD", "genetic", "5TGD")
]
result = find_physical(network)
print result
Example. Functions can call other functions. Let’s write two functions:
def read_fasta(path):
"""Reads a FASTA file with one-line sequences."""
fasta = {}
for line in open(path).readlines():
line = line.strip()
if line[0].startswith(">"):
header = line
else:
fasta[header] = line
return fasta
def compute_histogram(sequence):
"""Computes the histogram of the characters."""
histogram = {}
for letter in sequence:
if not histogram.has_key(letter):
histogram[letter] = 0
histogram[letter] += 1
return histogram
These functions can be used to implement a complex program that:
- Reads a FASTA file into a dictionary
- For each sequence in the FASTA file, computes the histogram of its letters
- Prints each sequence header and the corresponding histogram
as follows:
path = raw_input("enter a path: ")
fasta = read_fasta(path)
for header, sequence in fasta.items():
histogram = compute_histogram(sequence)
print "header =", header.lstrip(">"), ":"
print histogram
Example. Since functions can call other functions, the “call graph” of a program can become arbitrarily complicated. Let’s see a moderately realistic example of what a call graph looks like.
Let’s write a (mock!) program, composed of multiple functions, that asks the user for:
- the path to one or more FASTA files.
- the path to a file describing a physical protein interaction network (PIN).
and computes some average statistic (say, a histogram) of the amino acid composition of interacting proteins.
When ran, the program does the following:
- reads the sequence data from each FASTA file, see the
read_sequences()andread_fasta()functions - reads the interaction network with the
read_interactions()function - for each pair of interacting proteins, computes statistics about their
joint amino acid composition, through the
compute_aa_stats()function, and computes an “average” summary statistic in thecompute_avg_stats()function.
Here is the code:
def read_fasta(path):
"""Takes a path to a FASTA file, returns a
header->sequence dict."""
# TODO actually read the file
return "1A3A:A", "MANLFKLG..."
def read_sequences(paths):
"""Reads a bunch of FASTA files, returns a
list of dicts."""
header_to_seq = {}
for path in paths:
header, seq = read_fasta(path)
header_to_seq[header] = seq
return header_to_seq
def read_interactions(path):
"""Reads physical protein interactions from a
file. Returns a list of pairs of strings."""
# TODO actually read the file
return [("1A3A:A", "5AA3:F"), ("5AA3:F", "5K9C:A")]
def compute_aa_stats(seq1, seq2):
"""Compute amino acid statistics, e.g.
co-occurrence."""
# TODO actually compute co-occurrence and MI
cooccurrence = {"A": 0.2, "C": 0.01}
mutual_information = 0.72
return cooccurrence, mutual_information
def compute_avg_stats(sequences, interactions):
"""Takes a list of statistics (in some format) and
computes the average statistics."""
stats = []
for prot1, prot2 in interactions:
if not (sequences.has_key(prot1) and sequences.has_key(prot2)):
continue
seq1 = sequences[prot1]
seq2 = sequences[prot2]
stats.append(compute_aa_stats(seq1, seq2))
# TODO actually average all the collected statistics
return 0.3
def main():
"""The whole (fake) program."""
# Read the sequence files
paths = []
while True:
ans = raw_input("path to FASTA file: ")
if len(ans) == 0:
break
paths.append(ans)
sequences = read_sequences(paths)
# Read the interaction file
ans = raw_input("path to interaction data: ")
interactions = read_interactions(ans)
# Print the average stats
print "average stats =", compute_avg_stats(sequences, interactions)
main()
As you can see, Python begins by calling the main() function at the very
last line of the program. The main() function calls all the other
“major” functions: read_sequences(), read_interactions() and
compute_avg_stats().
The read_sequences() function internally calls the read_fasta()
function multiple times, once for each user-provided FASTA file.
The read_interactions() function calls no other function.
The compute_avg_stats() function uses the compute_aa_stats() function
to compute the statistics of individual protein-protein pairs.
The above can be summarized using a “call graph” like this:
Quiz. How many times is:
- the main() function called?
- the read_fasta() function called?
- the compute_aa_stats() function called?
Exercises¶
Given the function:
def function(arg): return arg
what is the type of the result of the following calls?
function(1)function({"1A3A": 123, "2B1F": 66})function([2*x for x in range(10)])function(2**-2)
Given the function:
def sum(a, b): return a + b
what is the type of the result of the following calls?
sum(2, 2)sum(range(10), range(10, 20))sum("I am a", "string")
Create a function
print_even_odd()that, given an integer, prints to screen"even"if the number is even, and"odd"otherwise.What is the result if we write:
result = print_even_odd(99) print result
Create a function
determine_even_odd()that takes an integer and returns the string"even"if the number is even, and the string"odd"otherwise.What is the result if we write:
determine_even_odd(99)
Create a function
check_alphanumeric()that takes a string and returnsTrueif the string is alphanumeric (contains only alphabetic or numeric characters) andFalseotherwise.To check whether a character is alphanumeric, use
inand the string"ABCDEFGHIJKLMNOPQRSTUVWXYZ0123456789".Hint. Lower case characters can be alphanumeric too!
Create a function
question()that doesn’t take any argument, asks the user for a file path and prints to screen the contents of the file. (Test the function with e.g. the filedata/aatable.)Create a function
wc()that takes a string and returns a triplet (tuple) of values:- The first element should be the length of the string.
- The second element should be the number of newline characters in the string.
- The third element should be the number of words (separated by spaces or newlines) in the string.
Create a function
print_dictionary()that takes a dictionary and prints to screen the key-value pairs of the dictionary, formatted as in the example below.Example: when applying
print_dictionary()to:dictionary = { "Arg": 0.7, "Lys": 0.1, "Cys": 0.1, "His": 0.1, }
a histogram of frequencies, the result should be:
>>> print_dictionary(dictionary) His -> 10.0% Cys -> 10.0% Arg -> 70.0% Lys -> 10.0%
Here the order in which lines are printed is not relevant.
As before, but keys should be ordered alphanumerically:
>>> print_ordered_dictionary(dictionary) Arg -> 70% Cys -> 10% His -> 10% Lys -> 10%
Hint: it would be convenient to extract dictionary keys, order them alphanumerically, then iterate on ordered keys printing each time the corresponding line.
Create a function
create_list_of_factorials()that takes an integern, and returns a list ofnelements.The
i-th element has to be the factorial ofi.For example:
>>> list = create_list_of_factorials(5) >>> print len(list) 5 # 5 elements, as requested >>> print list[0] 1 # the factorial of 0 >>> print list[1] 1 # the factorial of 1 >>> print list[2] 2 # the factorial of 2 >>> print list[3] 6 # the factorial of 3 >>> print list[4] 24 # the factorial of 4
Hint: it would be convenient to use the function
factorial()defined in one of the previous examples, to calculate the values of the list.Create a function
count_character()that takes two strings, the first representing text, the second representing a character.The function has to return the number of occurrences of the character in the text.
For example:
>>> print count_character("abbaa", "a") 3 # "a" appears 3 times >>> print count_character("abbaa", "b") 2 # "b" appears 2 times >>> print count_character("abbaa", "?") 0 # "?" never appears
Create a function
count_characters()that takes two strings, the first representing text, the second representing a set of characters.The function has to return a dictionary, where the characters to be found are the keys, and their occurrences in the text are the corresponding values.
For example:
>>> print count_characters("abbaa", "ab?") {"a": 3, "b": 2, "?": 0}
Create a function
distance()that takes two pairs of values(x1,y1)and(x2,y2)representing the two-dimensional coordinates of two points, and returns their Euclidean distance.Hint. Euclidean distance is calculated as \(\sqrt{(x1-x2)^2 + (y1-y2)^2}\)
Create a function
substring()that, given two strings, returnsTrueif the second is a substring of the first.Create a function
non_empty_substrings()that, given one string, returns a list of its non empty substrings.Create a function
count_substrings()that, given two stringshaystackandneedle, returns the number of occurrences ofneedleinhaystack.Create a function
longest_substring()that, given two strings, returns their longer common substring.Hint. You can solve this using the previous exercise!
Composing Functions into Programs¶
Exercises¶
Write a program that takes:
- a
pathto a PDB file, which describes the atomic structure of a multi-chain protein)
- a
two protein chain identifiers (
chain1andchain2)a distance threshold
threshold
The program should print the positions, coordinates, and distance of the atoms in the two chains whose Euclidean distance is closer than
threshold.Use the (modified) PDB file of insulin linked here to test your program.
Extract of the insulin PDB file:
# atom chain x y z # v v v v v ATOM 1 N GLY A 1 -8.932 16.943 14.339 1.00 15.82 N ATOM 2 CA GLY A 1 -9.595 17.000 13.020 1.00 13.75 C ATOM 3 C GLY A 1 -9.764 15.600 12.498 1.00 13.02 C ATOM 4 O GLY A 1 -9.598 14.645 13.247 1.00 11.89 O ... ATOM 10 N ILE A 2 -10.091 15.513 11.206 1.00 11.41 N ATOM 11 CA ILE A 2 -10.396 14.228 10.611 1.00 10.30 C ATOM 12 C ILE A 2 -9.248 13.293 10.677 1.00 9.25 C ATOM 13 O ILE A 2 -9.434 12.049 10.899 1.00 10.34 O ... ATOM 298 N ASN A 21 -12.001 17.750 -2.988 1.00 11.08 N ATOM 299 CA ASN A 21 -12.128 19.024 -3.668 0.58 13.56 C ATOM 300 CA ASN A 21 -11.958 19.077 -3.600 0.42 11.76 C ATOM 301 C ASN A 21 -13.435 19.106 -4.464 0.58 18.94 C ATOM 302 C ASN A 21 -13.065 19.211 -4.640 0.42 15.32 C ATOM 303 O ASN A 21 -13.993 18.044 -4.817 0.58 17.75 O ATOM 304 O ASN A 21 -14.121 18.561 -4.467 0.42 16.85 O ... ATOM 327 N PHE B 1 -21.703 1.132 3.507 1.00 14.54 N ATOM 328 CA PHE B 1 -20.263 1.256 3.871 1.00 11.12 C ATOM 329 C PHE B 1 -20.090 1.071 5.370 1.00 12.49 C ATOM 330 O PHE B 1 -21.062 1.257 6.129 1.00 20.68 O ...
Python: Functions (Solutions)¶
Solution: the same type of the value I pass to the function! The function returns the value of the argument without any change.
- an integer.
- a dictionary.
- a list.
- a rational.
Solution: the sum, or concatenation, of the two arguments, therefore:
- an integer.
- a list.
- a string.
Solution:
def print_even_odd(number): if number % 2 == 0: print "even" else: print "odd" print_even_odd(98) print_even_odd(99)
Note that
print_even_odd()already prints to screen, it’s not necessary to write:print print_even_odd(99)
otherwise Python will print:
even None
The
Nonecomes from the additionalprint. Sinceprint_even_odd()doesn’t have areturn, its result will always beNone. The additionalprintprints exactly thisNone.Solution:
def determine_even_odd(number): if number % 2 == 0: return "even" else: return "odd" print determine_even_odd(98) print determine_even_odd(99)
In this example, since there is no
printinsidedetermine_even_odd(), it’s necessary to add an additionalprintin order to print the result.Solution:
def check_alphanumeric(string): alphanumeric_chars = "ABCDEFGHIJKLMNOPQRSTUVWXYZ0123456789" alphanumeric_flag = True for character in string: if not character.upper() in alphanumeric_chars: alphanumeric_flag = False return alphanumeric_flag # Let's test the function print check_alphanumeric("ABC123") print check_alphanumeric("A!%$*@")
We can also use
breakto interrupt theforcycle as soon as we find a non-alphanumeric character (once we find a non-alphanumeric character, it is impossible thatalphanumeric_flagreturns toTrueagain):def check_alphanumeric(string): alphanumeric_chars = "ABCDEFGHIJKLMNOPQRSTUVWXYZ0123456789" alphanumeric_flag = True for character in string: if not character.upper() in alphanumeric_chars: alphanumeric_flag = False break # <-- exit from the cycle return alphanumeric_flag # Let's test the function print check_alphanumeric("ABC123") print check_alphanumeric("A!%$*@")
Alternatively, since upon executing
breakwe directly arrive toreturn, we can skip a step and write directlyreturn:def check_alphanumeric(string): alphanumeric_chars = "ABCDEFGHIJKLMNOPQRSTUVWXYZ0123456789" for character in string: if not character.upper() in alphanumeric_chars: # upon meetin a not alphanumeric character, # we can answer False return False # we arrive to the end of the for cycle only if `return` was never executed, # this happens only if the `if` condition has always been False for all characters # this means that all characters were alphanumeric # we can answer True return True # Let's test the function print check_alphanumeric("ABC123") print check_alphanumeric("A!%$*@")
Solution:
def question(): path = raw_input("write a path: ") print open(path).readlines() # Let's test the function question()
Solution:
def wc(string): num_chars = len(string) num_newlines = string.count("\n") num_words = len(string.split()) # split splits upon both spaces and newlines return (num_chars, num_newlines, num_words) # Let's test the function print wc("I am\nan awesome\nstring")
Solution:
def print_dictionary(a_dictionary): # here the printing order doesn't matter, # we can use the natural order provided by # `items()` for key, value in a_dictionary.items(): print key, "->", (str(value * 100.0) + "%") # Let's test the function dictionary = { "Arg": 0.7, "Lys": 0.1, "Cys": 0.1, "His": 0.1, } print_dictionary(dictionary)
Solution:
def print_ordered_dictionary(a_dictionary): # extract and order keys ordered_keys = a_dictionary.keys() ordered_keys.sort() # now let's print key-value pairs in the correct order for key in ordered_keys: value = a_dictionary[key] print key, "->", (str(value * 100.0) + "%") # Let's test the function dictionary = { "Arg": 0.7, "Lys": 0.1, "Cys": 0.1, "His": 0.1, } print_ordered_dictionary(dictionary)
Solution:
# taken from the example def factorial(n): fact = 1 for k in range(1, n+1): fact = fact * k return fact def create_list_of_factorials(n): list_of_factorials = [] for i in range(n): list_of_factorials.append(factorial(i)) return list_of_factorials # Let's test the function print create_list_of_factorials(2) print create_list_of_factorials(4) print create_list_of_factorials(6)
here we reused the function
factorial()defined in a previous example.Solution:
def count_character(text, search_char): counts = 0 for character in text: if character == search_char: counts += 1 return counts # Let's test the function print count_character("abbaa", "a") print count_character("abbaa", "b") print count_character("abbaa", "?")
or, more simply, we can use
count():def count_character(text, search_char): return text.count(search_char) # Let's test the function print count_character("abbaa", "a") print count_character("abbaa", "b") print count_character("abbaa", "?")
Solution:
def count_characters(text, search_chars): counts = {} for search_char in search_chars: counts[search_char] = count_character(text, search_char) return counts # Let's test the function print count_characters("abbaa", "ab?")
Here, we reused the function defined in the previous exercise.
Solution:
def distance(pair1, pair2): x1, y1 = pair1 x2, y2 = pair2 dx = x1 - x2 dy = y1 - y2 return (float(dx)**2 + float(dy)**2)**0.5 # Let's test the function print distance((0, 0), (1, 1)) print distance((2, 3), (3, 2))
Solution:
def substring(first, second): return second in first # Let's test the function print substring("ACGT", "T") print substring("ACGT", "x")
Solution:
def non_empty_substrings(input_string): substrings = [] # all the possible positions to start a substring for start in range(len(input_string)): # all the possible positions to end a substring for end in range(start + 1, len(input_string) + 1): # extract the substring and update the list substrings.append(input_string[start:end]) return substrings # Let's test the function print non_empty_substrings("ACTG")
Soluzione:
def count_substrings(haystack, needle): occurrences = 0 for start in range(len(haystack)): for end in range(start+1, len(haystack)+1): # to be sure, we print the substring print start, end, ":", haystack[start:end], "==", needle, "?" # check whether the substring is equal to `needle` if haystack[start:end] == needle: print "here is an occurrence!" occurrences += 1 return occurrences # Let's test the function print count_substrings("ACTGXACTG", "ACTG")
Solution:
def longest_substring(string1, string2): # let's reuse the previous function substrings1 = non_empty_substrings(string1) substrings2 = non_empty_substrings(string2) # find the longest common substring longest_sub = "" for substring1 in substrings1: for substring2 in substrings2: if substring1 == substring2 and \ len(substring1) > len(longest_sub): # aggiorno longest_sub = substring1 return longest_sub # Let's test the function print longest_substring("ACTG", "GCTA") print longest_substring("", "ACTG") print longest_substring("ACTG", "")
Python: Programs¶
Exercises¶
Write a Python program that:
- takes as input a file name
filenamecontaining the multiple alignment of a protein sequence - prints, for each sequence position, the corresponding alignment profile, i.e how many times each amino acid (considering also gaps as an option) was found in that position of the alignment.
Input example (copy-and-paste into a file):
0.012 C ---------CCAHSLVVTEASAD------------------------------------------------- 0.013 G ---------GGGYFAVFATAFSS------------------------------------------------- 0.024 V ---------VILINLLLYVIIIE-------VI---------------------------------------- 0.014 P ---------PPPCKRGKG-SEDS-------RV---------------------------------------- 0.013 A ---------AKACLSSVY-VIN--------FT------L--------------------------------- 0.014 I ---------IIPPEY-QLLVFI--------HL------P--------------------------------- 0.019 Q ---------QEKENEEGTQGGE--------LG------L--------------------------------- 0.020 P ---------PPTPPSSQKP-DI--------SV------P------A-------------------------- 0.077 V --N------VKAP-PPREESTPVGAVKGTCR-------P--L---SR-E----L-DC---AE-W-S---MQ- 0.087 L --A------LLLP-VISESPKEDHSPPPSSP-------L--E---FD-IG---WDRG---EA-T-T---QI- 0.106 S --K------SPPL-GSEERIPVTSSSGETGF-------N--KA--ADDQT-F-RLLW--KHP-G-P---TQ- 0.103 G --T------GGQY-V-PIQKPDNIRFFQTSF-------DG-LT--DSRGA-Y-TDSD--AVD-R-Q---VA- 0.202 L --F------LAA--RQLQSDSSGPATEMSHG-QQQSSSRD-SGQSKTRNANV-HQMPV-LRGLH-Q--NEGS 0.274 S VVI-E-PPTSNE--EFYSSNVLLPKGGGPSA-GGGFLSEGPGEENPNSDSGR-ASASGPKSFAP-P-DENQT 0.399 R GGR-R-RRRRRRR-QKRRRRKER-RFRRTRR-RRRATSRKKRERGDMRRERS-RRLRRDELETR-SAALKDW 0.610 I IVI-I-IIIIIIM-IIVIVIIIVIIVIVRII-IIIILIIIIIIIIVIIIIII-IIIIVHYVITI-IYLVIVL 0.463 V YAV-V-VIVVVVAVLLIVIVVVVIVIIVIIV-LMAHQRVVVVIVYAVRVVVV-IIIVAINMVVYPISHLRII 0.506 N GGG-G-GGGNGGYEGGNGNSGNNEGNNGNCG-GGGGGNGNGGNGNNGEGGGG-GGGNDANGGSNLGSRGHGR 0.794 G GGG-G-GGGGGGGAGGGGGGGGGGGGGGGGG-GGGGDGGGGGGGGGGGGGGG-GGGGGGSGGTGGGSGGGGS 0.288 E HHQ-S-HHKESITSHHKSRSSQQTDTHSGQS-EEETRVSTSEKVVGMQ-RIH-TYTVS-YGQVIK-SITPVP 0.323 E DED-Y-PVEEDEASNNPSDDPENEEEENQNV-DDDKKRNTEVKEEDKL-RDA-EDDED-LTKIKE-PPELSV 0.493 A IIA-I-SSTAASANAAASAAAVAAAAAARAAAVAAIITAAATAVAAVARAAS-AVVTA-PVASTCSAVVTAQ 0.320 V SPN-P-DSTVVTRLDPDSPKRVNNAEEAVKSEADVDERSGEPKPKADKPRVLMNDEER-EPQFTPNRQPSNH 0.351 P IIIII-VIIPPIPVIVLVPLLPRLPPKVNKEVQALIIIPPRKKIFEIDSPAPTIIDIR-NVFRVPAIQVIDR 0.372 G ESQEE-WKSGHNNHKAFGGGHHGHHHGNENGGGTGKTYGGNFNHDNEDGHGGRSEGNGTFGGPEHTEQGVIT 0.388 S QDDQY-HEESSQEQLSSQSQQSQEESESTSQSEAQDDLDEEQSLFSQEGADAHNDKEQTNARRSSSDNKKIE 0.347 W AAYAV-QEVWYWYNHANFFFFIFFFAAAVAFQYTLAAAAFMYFTWFVFYWAAWFAAFFPVKHYLRGYYYYAF 0.739 P PPPPP-PKPPPSPPPPPPKPPPPPPPPPPPPPPTPPPPVPPPQPTPPTSPPPPPPPPPPPTRPGPPPGPPRP 0.521 W FFYWW-HYYWWSWWWWWYYWWYYYYYFYFYHHWFYWWYYFYWYWFYYGFFWYWWYYMYYYYYYGWWSYFFSF
Execution example:
python program.py Data file: alignment el - A C D E F G H I K L M N P Q R S T V W X Y C 58 3 2 1 1 0 0 1 0 0 1 0 0 0 0 0 2 1 2 0 0 0 G 58 3 0 0 0 3 3 0 0 0 0 0 0 0 0 0 2 1 1 0 0 1 V 56 0 0 0 1 0 0 0 6 0 4 0 1 0 0 0 0 0 3 0 0 1 P 57 0 1 1 1 0 2 0 0 2 0 0 0 3 0 2 2 0 1 0 0 0 A 57 2 1 0 0 1 0 0 1 1 2 0 1 0 0 0 2 1 2 0 0 1 I 57 0 0 0 1 1 0 1 3 0 3 0 0 3 1 0 0 0 1 0 0 1 Q 56 0 0 0 5 0 4 0 0 1 2 0 1 0 2 0 0 1 0 0 0 0 P 56 1 0 1 0 0 0 0 1 1 0 0 0 6 1 0 3 1 1 0 0 0 V 36 3 2 1 4 0 2 0 0 2 2 1 1 5 1 3 3 2 3 1 0 0 ....
Warning
Note that not all the rows contain all the possible aminoacids and gaps (see all the 0 values present in the table). In order to account for this, it’s necessary to extract an alphabet (you can see it in the first row of the output) with all the possible alternatives, to be used as a reference.
You can use five separate functions:
- A function reading the file
filenameand returning a list of couples residue-alignment - A function taking as input the list read from the file, and returning an alphabetically ordered list of all the possible characters appearing in the alignment, to be used as alphabet
- A function taking as input a string with an alignment (in a position) and returning a profile (dictionary from character to the corresponding number of occurrences in the alignment)
- A function taking as input the list read from the file and the alphabet, printing a header (see execution example) and, for each position in the list, the corresponding sequence element and the profile (obtained from function 3) ordered as alphabet (if the letter is not present in the profile, the number of occurrences will be 0)
- A function that runs the program using the functions defined above.
- takes as input a file name
Write a Python program that:
- takes as input a file name
filenamecontaining the annotation of a protein sequence in UNIPROT format - prints, for each secondary structure element (HELIX, STRAND, TURN), the average length of the element, in amino acids, and the average number of consecutive times the element occurs in the sequence
Input example (protein Q1NZL3 on Uniprot):
ID ZN224_HUMAN Reviewed; 707 AA. AC Q9NZL3; A6NFW9; P17033; Q86V10; Q8IZC8; Q9UID9; Q9Y2P6; DT 08-DEC-2000, integrated into UniProtKB/Swiss-Prot. DT 30-NOV-2010, sequence version 3. .... .... .... FT {ECO:0000305}. FT CONFLICT 562 562 H -> R (in Ref. 1; AAF04106). FT {ECO:0000305}. FT STRAND 174 177 {ECO:0000244|PDB:2EN8}. FT TURN 179 181 {ECO:0000244|PDB:2EN8}. FT STRAND 184 187 {ECO:0000244|PDB:2EN8}. FT HELIX 188 195 {ECO:0000244|PDB:2EN8}. FT TURN 196 198 {ECO:0000244|PDB:2EN8}. FT STRAND 207 209 {ECO:0000244|PDB:2EM6}. FT HELIX 216 223 {ECO:0000244|PDB:2EM6}. FT TURN 224 226 {ECO:0000244|PDB:2EM6}. FT STRAND 227 229 {ECO:0000244|PDB:2EM6}. FT STRAND 235 237 {ECO:0000244|PDB:2ELY}. FT STRAND 241 243 {ECO:0000244|PDB:2ELY}.
Execution example:
python program.py Data file: Q9NZL3.txt average lengths {’TURN’: 3.125, ’HELIX’: 7.285714285714286, ’STRAND’: 3.323529411764706} average number of consecutive occurrences {’TURN’: 1.0, ’HELIX’: 1.4, ’STRAND’: 1.736842105263158}You can use five separate functions:
- A function reading the file
filenameand returning a list of pairs: secondary structure element (ss) - length. - A function taking as input the list read from the file, and returning two dictionaries, both having ss as keys, the first having as value the corresponding sum of lengths, the second the number of times the ss element occurs.
- A function taking as input two dictionaries and returning a new dictionary obtained by normalizing the values of the first with the values of the second.
- A function taking as input the list read from the file, and returning two dictionaries, both having ss as keys: the first dictionary maps from ss the number of occurrences; the second maps from ss to the number of non-consecutive occurrences. The function will return the normalized dictionary of occurrences.
- A function that implements the program using the above functions.
- takes as input a file name
Write a Python program that:
- Takes a
pathto a file describing a list of donor (D) splicing sites in FASTA format. Each site contains 15 base pairs: 7 before the site, 8 after the site. - An integer
thresholdin between 2 and 8.
The program should:
Print the number of occurrences of the sub-sequences (1) before and (2) after the donor site, for all sub-sequences of length in-between 2 and 7 (before the site) or 8 (after the site). Sub-sequences occurring less than
thresholdtimes should not be printed.The number of occurrences should be computed over all the sequences contained in the FASTA file.
The full input file is here:
Example input
>HUMGLUT4B_2257 <-- header CTCCGAAGTAGGATT <-- site sequence (7 before + 8 after) >HUMGLUT4B_3651 TCAGAAGGTGAGGGC >HUMGLUT4B_3935 TTGGAAGGTTCGCAG >HUMGLUT4B_4152 TACTCAGGTACTCAC >HUMGLUT4B_4379 CGCCCAGGTGACCGG >HUMGLUT4B_4669 AGAAAGAGTAAGCTC ...
For instance, given the sequence:
CTCCGAAGTAGGATTthe sequence after the site is
GTAGGATT, and its sub-sequences are:GT, GTA, GTAG, ..., GTAGGATT
while the sequence before the site is
CTCCGAA, and its sub-sequences are:AA, GAA, CGAA, ..., CTCCGAA
The expected result with
threshold = 100is:> python splice_patterns.py Insert path: splice_donor.fasta Insert threshold: 100 Most frequent sub-sequences before the site: {'AAG': 208, 'AG': 583, 'GG': 107, 'GAG': 108, 'TG': 135, 'CAG': 247} Most frequent sub-sequences after the site: {'GTA': 568, 'GT': 1116, 'GTAAGT': 126, 'GTGAG': 388, 'GTG': 491, 'GTGA': 399, 'GTGAGT': 204, 'GTAA': 379, 'GTAAG': 300, 'GTGAGTG': 116}
meaning that
"AAG"appears208times before the site,"AG"appears583times before the site, ...,"GTA"appears568times after the site,"GT"appears1116times after the site, etc.Note
We suggest implementing five functions:
- A function that reads the input file and returns two collections: a collection of all sequences before the site, and a collection of all sequences after the site.
- A function that takes a collection of sequences after the site, and returns a dictionary mapping from sub-sequences to number of occurrences.
- A function that takes a collection of sequences before the site, and returns a dictionary mapping from sub-sequences to number of occurrences. (note that sub-sequences start at the end of the sequence of the file, and grow backwards.)
- A function that takes a dictionary of counts and a threshold, and returns a dictionary of counts with only the key-value pairs whose value is larger than or equal to the threshold.
- A function that implements the program using the above.
- Takes a
Write a Python program that:
- Takes a
pathto a file describing the association between proteins and the PFAM domains that appear in the protein. - Prints the list of all the PFAM domains (including their ID and name), sorted by number of occurrences, as well as the number of proteins that each PFAM domain appears into.
The full input file is output_pfam.
Example input:
78184355|ref|YP_376790.1| 28 145 PF01475.11 1 124 ls 62.1 2.1e-15 FUR 78185596|ref|YP_378030.1| 31 111 PF00575.15 1 79 fs 33.4 2.8e-08 S1 78185596|ref|YP_378030.1| 113 381 PF10150.1 1 273 ls 436.7 3.5e-128 RNase_E_G 78185773|ref|YP_378207.1| 70 222 PF04087.6 1 147 ls 221.5 2.1e-63 DUF389 78183836|ref|YP_376270.1| 13 375 PF01266.16 1 399 ls 182.5 1.2e-51 DAO 78184892|ref|YP_377327.1| 5 191 PF00702.18 1 184 fs 99.1 5.5e-28 Hydrolase 78183858|ref|YP_376292.1| 78 482 PF00909.13 1 455 ls 519.6 4.1e-153 Ammonium_transp ... ^^^^^^^^^^^^^^^^^^^^^^^^^ ^^^^^^^^^^ ^^^^^^^^^^^^^^^ protein domain ID domain name
The expected result is:
> python pfam_statistics.py Type path: output_pfam PFAM ID PFAM Name NumOcc NumDiffProt PF00005.19 ABC_tran 35 32 PF00132.16 Hexapep 29 5 PF00534.12 Glycos_transf_1 21 20 PF00515.20 TPR_1 17 8 PF01370.13 Epimerase 16 16 PF03130.8 HEAT_PBS 15 6 PF04055.13 Radical_SAM 14 14 PF00502.11 Phycobilisome 13 12 PF00427.13 PBS_linker_poly 13 10 PF00271.23 Helicase_C 13 13 PF00528.14 BPD_transp_1 12 11 PF00004.21 AAA 12 12 PF00805.14 Pentapeptide 11 5 PF00535.18 Glycos_transf_2 11 11 PF00353.11 HemolysinCabind 11 3 PF07862.3 Nif11 10 10 PF01926.15 MMR_HSR1 10 9
The output shows that, for instance, the domain with ID
PF00005.19and nameABC_tranoccurs35different times in the complete file, and in particular it occurs in32distinct proteins.Warning
The output text columns are not required to be aligned.
Note
We suggest implementing five functions:
- A function that reads the input file and returns a dictionary mapping from domain PFAM ID to doman PFAM name, and for each protein the list of its domains.
- A function that takes the information read from the file, and returns a dictionary from domains to their number of occurrences.
- A function that takes the information read from the file, and returns a dictionary from domains to their number of distinct proteins they appear into.
- A function that print the output as shown in the expected results. The domains should be sorted by their total number of occurrences.
- A function that implements the program using the above.
- Takes a
Write a Python program that:
- Takes a
pathto a file containing protein sequences in FASTA format. The header of the FASTA contains information about the cellular localization and the organism of the protein. - Prints, for each cellular localization, the number of proteins, divided by organism, and sorted from the most frequent to the less frequent organism.
The full input file is here:
Example input:
>7B2_HUMAN:Secretory MVSRMVSTMLSGLLFWLASGWTPAFAYSPRTPDRVSEADIQRLLHGVME ... >A1AG1_MUSCR:Secretory MALHMILVMLSLLPLLEAQNPEHVNITIGEPITNETLSWLSDKWFFIGA ... >A1BG_HUMAN:Secretory MSMLVVFLLLWGVTWGPVTEAAIFYETQPSLWAESESLLKPLANVTLTC ... ... >ZNF22_RAT:Nucleus MRLGKPQKGGISRSATQGKTYESRRKTARQRQKWGVAIRFDSGLSRRRR ... >ZNF42_HUMAN:Nucleus MRPAVLGSPDRAPPEDEGPVMVKLEDSEEEGEAALWDPGPEAARLRFRC ...
The expected result is:
> python program.py Name of file: sequences.fasta Mitochondrion 107:HUMAN 27:BOVIN 18:DROME 16:MOUSE 7:RAT 3:ASCSU ... Secretory 191:HUMAN 79:MOUSE 59:DROME 35:BOVIN 30:RAT 19:CHICK ... Nucleus 636:HUMAN 188:DROME 145:MOUSE 65:CAEEL 27:RAT ... Cytoplasm 203:HUMAN 72:MOUSE 47:DROME 30:RAT 20:CAEEL 13:BOVIN ...
Note
We suggest implementing four functions:
- A function that reads the input file and returns a dictionary mapping from each cellular localization to the list of protein with that localization
- A function that takes the dictionary generated by the previous function and returns a new dictionary, mapping from cellular localizations to dictionaries of counts (mapping from organism to number of proteins belonging to the organism)
- A function that takes the dictionary generated by the previous function and, for each cellular localization, prints the list of counts, ordered by decreasing number of occurrences.
- A function that implements the program using the above functions.
- Takes a
Write a function
findPerfectMatches(filename,pattern)that:- Takes the name of a file
filenamecontaining a list of RNA sequences, and an RNA string (for example, a microRNA sequence). - Prints, for each sequence, the name and the list of positions corresponding to perfect matches (by complementarity) with the string (only for sequences with at least one match).
The expected result is:
>>> import utility >>> utility.findPerfectMatches(’utr.txt’,’acgaatt’) ENSG00000050344 [1186] ENSG00000109929 [204, 373, 3336] ENSG00000155850 [2162, 5387] ENSG00000073756 [1152] ENSG00000175445 [693] ENSG00000159167 [781] ENSG00000138061 [1229] ENSG00000152268 [1362] ENSG00000197121 [3024] ENSG00000115159 [1111] ENSG00000169908 [1695] ENSG00000150938 [1751] ENSG00000179314 [2782] ENSG00000075223 [1743]
Note
We suggest implementing five functions:
- A function that reads the input file and returns a dictionary mapping from name to sequence.
- A function that takes as input a string and returns the complementary sequence (note that sequences and strings have T, and not U, even if they represent RNA sequences).
- A function that takes two sequences and return the list of occurrences of the second string on the first one (look carefully at the help of the function
find). - A function taking the dictionary of sequences and the converted string, that calculates for each sequence the match positions, using the previous function, and if there is at least one match, prints the name of the sequence and the list of matches.
- A function that implements the program using the above.
- Takes the name of a file
Midterm exam 2016/11/02:
- Given:
- The
hierarchy.txtfile. - Optionally, a pair of protein identifiers.
- The
- write a Python program that:
- If the two protein identifiers are given, prints the list of domains shared by the two proteins; further, for each shared domain, it prints the residue histogram of their sequences.
- If no protein identifiers are given, it prints the above information for all pairs of proteins. The output text columns are not required to be aligned.
Warning
Same domains appear in more than one protein; different occurrences of a domain however may have different sequences!
Example input:
# Protein Domain Position Residue YAL003W PF00736 00120 W YAL003W PF00736 00121 I YAL003W PF00736 00154 Q YAL038W PF00224 00077 E YAL038W PF00224 00236 K YAL038W PF00224 00327 H YAL038W PF00224 00362 I YAL038W PF02887 00391 L YAL038W PF02887 00415 S YAL038W PF02887 00432 G YAL038W PF02887 00441 F YAL038W PF02887 00450 M ...
Expected output:
$ python commdomains.py insert the path to the hierarchy file: hierarchy.txt all pairs? (Y or N) N insert 1st protein ID: YIL109C insert 2nd protein ID: YPR181C YIL109C YPR181C shared domains: ['PF00626', 'PF04810', 'PF04811'] PF00626 histogram = {'I': 1, 'R': 1, 'T': 1, 'W': 3, 'V': 1} PF04810 histogram = {'S': 1, 'R': 1} PF04811 histogram = {'A': 1, 'E': 1, 'D': 3, 'G': 4, 'F': 3} PF04815 histogram = {'C': 1, 'D': 2, 'I': 1, 'Q': 2, 'P': 2} PF08033 histogram = {'C': 1, 'E': 2, 'D': 1, 'I': 3, 'L': 2}Note
We suggest implementing five functions:
- A function that takes a path to the hierarchy file, and returns
a map from each protein to a dictionary; this dictionary maps
from each domain in the protein to its sequence. Picture:
sequence = hierarchy[protein][domain] - A function that takes two lists and returns their intersection.
- A function that takes a sequence and returns the histogram of its residues.
- A function that takes the hierarchy read from the file and two protein identifiers, and prints the shared domains. Then, for every shared domain, it extracts the domain sequences from the two proteins, and prints their histogram.
- A function that implements the program.
Python: Programs (Solutions)¶
Solution (one among many):
from pprint import pprint def readFile(filename): f = open(filename) couples = [] for l in f.readlines(): words = l.strip().split() couples.append([words[1], words[2]]) f.close() return couples def extractAlphabet(data): sequences = [] for element in data: sequences += element[1] chars = "".join(sequences) alphabet={} for c in chars: alphabet[c]=1 alphabet=alphabet.keys() alphabet.sort() return alphabet def computeProfile(sequence): profile={} for el in sequence: if not profile.has_key(el): profile[el] = 1 else: profile[el] += 1 return profile def printProfiles(data,alphabet): print "el " + " ".join(alphabet) for row in data: profile = computeProfile(row[1]) print_line = [str(row[0])] for l in alphabet: if profile.has_key(l): print_line.append(str(profile[l])) else: print_line.append("0") print(" ".join(print_line)) def main(): couples = readFile("alignment.txt") alphabet = extractAlphabet(couples) printProfiles(couples,alphabet) return main()
Solution (one among many):
def readFile(filename): f=open(filename) struct=[] for row in f: if not row.startswith("FT"): continue data = row.split() if data[1] == 'STRAND' or data[1] == 'HELIX' or data[1] == 'TURN': struct.append((data[1],int(data[3])-int(data[2])+1)) f.close() return struct def computeAverageLengths(struct): lengths={} num={} for (ss,length) in struct: if not lengths.has_key(ss): lengths[ss]=length num[ss]=1 else: lengths[ss]+=length num[ss]+=1 return lengths,num def computeAverageConsecutive(struct): durations={} num={} ss=struct[0][0] dur=1 for i in range(1,len(struct)): if struct[i][0] == ss: dur+=1 else: if not durations.has_key(ss): durations[ss] = dur num[ss]=1 else: durations[ss] += dur num[ss]+=1 ss=struct[i][0] dur=1 durations[ss] += dur num[ss]+=1 return durations,num def normalize(values,sizes): normvalues={} for (k,v) in values.items(): normvalues[k] = float(values[k]) / sizes[k] return normvalues def computeSecondaryStructureStats(filename): struct=readFile(filename) print "average lengths " l1, l2 = computeAverageLengths(struct) print normalize(l1,l2) print "average number of consecutive occurrences " o1, o2 = computeAverageConsecutive(struct) print normalize(o1,o2) filename=raw_input("Data file: ") computeSecondaryStructureStats(filename)
Solution (one among many):
def load_sites(filename): f = open(filename) pre_sites=[] post_sites=[] for row in f: if row[0] == '>': continue pre_sites.append(row[:7]) post_sites.append(row[7:]) f.close() return pre_sites,post_sites def compute_post_site_patterns(sites, maxlen): patterns={} for site in sites: for k in range(2,maxlen): pattern = site[:k] if not patterns.has_key(pattern): patterns[pattern]=1 else: patterns[pattern]+=1 return patterns def compute_pre_site_patterns(sites, maxlen): patterns={} for site in sites: for k in range(2,maxlen+1): pattern = site[-k:] if not patterns.has_key(pattern): patterns[pattern]=1 else: patterns[pattern]+=1 return patterns def filter_by_freq(freqs, threshold): filtered_freqs={} for (k,v) in freqs.items(): if v >= threshold: filtered_freqs[k]=v return filtered_freqs def splice_patterns(filename, threshold): pre_sites,post_sites = load_sites(filename) print filter_by_freq(compute_pre_site_patterns(pre_sites, 7), threshold) print filter_by_freq(compute_post_site_patterns(post_sites, 8), threshold) filename=raw_input("Nome file: ") threshold=int(raw_input("Soglia: ")) splice_patterns(filename, threshold)
Solution (one among many):
def read_file(filename): f=open(filename) domains={} f.readline() for row in f.readlines(): data=row.split() domain=data[8] if not domains.has_key(domain): domains[domain]=[] domains[domain].append((data[0],int(data[1]),int(data[2]))) return domains def compute_average_length(domains): length4domain={} for (domain,entries) in domains.items(): avg_length=0. for (prot,start,end) in entries: avg_length+=end-start+1 avg_length/=len(entries) length4domain[domain]=avg_length return length4domain def compute_number_of_proteins(domains): prot4domain={} for (domain,entries) in domains.items(): prots={} for (prot,start,end) in entries: prots[prot]=1 prot4domain[domain]=len(prots) return prot4domain def print_statistics(length4domain,prot4domain): print "domain\t\t\tavg_length\tnum_prots" for domain in length4domain.keys(): print "%s\t\t\t%f\t%d" %(domain,length4domain[domain],prot4domain[domain]) def compute_domain_statistics(filename): domains = read_file(filename) lengths=compute_average_length(domains) prots=compute_number_of_proteins(domains) print_statistics(lengths,prots) filename=raw_input("Name of file: ") compute_domain_statistics(filename)
Solution (one among many):
def readFile(filename): f=open(filename) seqs={} for row in f: if row[0] == '>': data=row[1:].strip().split(':') name,localization=data[0],data[1] if not seqs.has_key(localization): seqs[localization]=[] seqs[localization].append(name) return seqs def countorganism_by_localization(prots): orgbyloc={} for (localization,data) in prots.items(): orgs={} for name in data: org = name.split("_")[1] if not orgs.has_key(org): orgs[org]=1 else: orgs[org]+=1 orgbyloc[localization]=orgs return orgbyloc def print_counts(counts_by_loc): for localization,counts in counts_by_loc.items(): print localization countlist=[(count,org) for (org,count) in counts.items()] countlist.sort() countlist.reverse() print " ".join(["%d:%s" %(count,org) for (count,org) in countlist]) def compute_organismcount_by_localization(filename): prots=readFile(filename) counts_by_loc=countorganism_by_localization(prots) print_counts(counts_by_loc) filename=raw_input("Name of file: ") compute_organismcount_by_localization(filename)
Solution (one among many):
def loadRNA(filename): f=open(filename) rna={} for row in f: data=row.split() rna[data[0]]=data[1] f.close() return rna def findMatches(seq,subseq): matches=[] start=0 while start != -1: start=seq.find(subseq,start) if start > -1: matches.append(start) start+=1 return matches def findAllMatches(rna,subseq): for (name,seq) in rna.items(): matches=findMatches(seq,subseq) if matches: print name,matches def complementary(pattern): comp={"a" : "t", "t" : "a", "c" : "g", "g" : "c"} return "".join([comp[p] for p in pattern]) def findPerfectMatches(filename,pattern): rna=loadRNA(filename) comppattern=complementary(pattern) findAllMatches(rna,comppattern) findPerfectMatches('utr.txt','acgaatt')
Midterm exam solution (one among many):
def read_hierarchy(path): fp = open(path) hierarchy = {} for line in fp.readlines()[1:]: prot, dom, res, aa = line.strip().split() if not hierarchy.has_key(prot): hierarchy[prot] = {} if not hierarchy[prot].has_key(dom): hierarchy[prot][dom] = [] hierarchy[prot][dom].append(aa) fp.close() return hierarchy def intersect(list1, list2): intersection = [] for el1 in list1: if el1 in list2: intersection.append(el1) intersection.sort() return intersection def histogram(seq): counts = {} for char in seq: if not counts.has_key(char): counts[char] = 0 counts[char] += 1 return counts def print_shared_domain_stats(data, pi, pj): domainsi = data[pi].keys() domainsj = data[pj].keys() shared = intersect(domainsi, domainsj) print pi, pj, "shared domains:", shared if len(shared) == 0: return for domain in shared: seqi = "".join(data[pi][domain]) seqj = "".join(data[pj][domain]) print domain, "histogram =", histogram(seqi + seqj) def run(): path = raw_input("insert the path to the hierarchy file: ") hierarchy = read_hierarchy(path) if raw_input("all pairs? (Y or N) ") == "N": p1 = raw_input("insert 1st protein ID: ") p2 = raw_input("insert 2nd protein ID: ") print_shared_domain_stats(hierarchy, p1, p2) else: proteins = hierarchy.keys() proteins.sort() for i in range(len(proteins)): for j in range(i + 1, len(proteins)): print_shared_domain_stats(hierarchy, proteins[i], proteins[j]) run()
External Modules¶
Importing Packages¶
Packages are just Python modules that offer additional functionality. There are many packages in the wild, take a look at the Python Package Index for an overview.
In order to make use of a package, you have to first import it:
import somepackage
Once imported, you can perform the usual operations on the package, e.g.:
>>> help(somepackage)
>>> print somepackage.__version__
>>> somepackage.function()
The Python Standard Library is installed by default together with Python 2 (and Python 3). It provides a lot of packages for dealing with many different tasks, e.g. regular expressions (more on that later).
Other specific packages do not come along with Python 2 (or 3) by default, so they have to be installed separately.
Warning
If you try to import a package, and get:
>>> import iamnotinstalled
Traceback (most recent call last):
File "<stdin>", line 1, in <module>
ImportError: No module named iamnotinstalled
it means that the package is not installed on your machine.
Luckily, it is easy to install a package with Python. The process is very streamlined. If you want to install a new package, just type (from the shell):
$ pip install thepackageyouwanttoinstall --user
This will install the package named thepackageyouwanttoinstall into your
home directory (more specifically, inside the ~/.local/lib/python2.7/
directory).
After the installation, Python will automatically pick up the package and allow you to import it.
Warning
These instructions assume that you have the pip installer.
This is the case for most GNU/Linux distributions and MacOS X versions
>= El Capitan.
If pip is not installed, either install it from your package manager
on GNU/Linux, brew on MacOS X, or use the generic setup script provided
by the Python Packaging Authority
Example. Let’s install the biopython package:
$ pip install biopython --user
The pip command will download the package (and all its dependencies) from
the internet, and install them in the correct order.
After pip is done, open a Python interpreter and try to import the
biopython package:
>>> import Bio
Warning
The pip installer and the python interpreter may use different
names to refer to the same package.
In the example above, the Biopython package is called biopython by
the pip installer, and simply Bio from within Python.
Please refer to the package documentation (i.e. its website) to figure out how it is called in the two different settings.
Example. Let’s say that you want to import the numpy module. Just
write:
import numpy
Once imported, you can take a peek at the various functionalty supported by
the numpy module by typing:
help(numpy)
For instance, to use the arccos function provided by the numpy module,
type:
print numpy.arccos(0)
You can also abbreviate the name of the package with a shorthand, as follows:
>>> import numpy as np
>>> print np.__version__
1.11.2
>>> np.arccos(0)
1.5707963267948966
>>> np.arccos(1)
0.0
Example. You can import specific sub-modules using the following notation:
import module.submodule
Then you can call the functions available in the sub-module as:
module.submodule.function("stuff")
For instance, to import the linalg (linear algebra) submodule of the
numpy package, write:
>>> import numpy.linalg
>>> help(numpy.linalg)
>>> help(numpy.linalg.eig)
Now, you can also import the submodule as a standalone package:
>>> from numpy import linalg
>>> help(linalg.eig)
as well as using the shorthand trick:
>>> import numpy.linalg as la
>>> help(la.eig)
or both:
>>> from numpy import linalg as la
>>> help(la.eig)
Finally, you can import individual functions:
>>> from numpy import arccos
>>> print arccos(0)
as well as multiple functions:
>>> from numpy import arccos, arcsin
>>> print arccos(0)
1.57079632679
>>> print arcsin(0)
0.0
Warning
The __future__ “module” is a special module used to import Python 3
functionality into Python 2 programs. It can be useful for writing
code compatible with both Python 2 and Python 3.
For instance, although print is a statement in Python 2, in Python
3 it becomes a function, meaning that our beloved:
print "stuff"
does not work anymore: in Python 3, print is a proper function an
requires brackets:
print("stuff")
In order to have a true print function in your Python 2 program,
use the following import at the very beginning of your script:
from __future__ import print_function
A useful feature to import is:
from __future__ import division
Once imported, the division feature makes division beteen integers
return a float if needed!
Exercises - Packages¶
Import some of the packages from the Python Standard Library. Some useful ones are:
The
mathpackage. It offers a plethora of mathematical utility functions. Use itsfactorial()function to compute the factorial of all integers from1to100, i.e.1!,2!, ...Remark. It is much faster than our naive implementation!
The
globpackage. It includes a methodglob()that takes a path to a directory, and returns the list of the contents of the directory, pretty much like thelsshell command.Use the
glob()function and print the list of the files in your home directory.Remark. Unfortunately
glob()does not understand the~path.The
time()package. It provides atime()function that returns the number of seconds since the Epoch, i.e. midnight UTC of 1st January 1970.It also provides a
sleep()function that takes a number of seconds, and makes your program pause for the specified amount of time (with reasonable approximation).After properly importing the
time()andsleep()functions from thetimepackage, check what this code does:t0 = time() for i in range(5): sleep(1) print "it took me {}s to get here".format(time() - t0) print "done in approx {}s".format(time() - t0)
The
pickle()package. It provides facilities for storing arbitrary Python data structures to disk, and loading them back.It is a must-have package for saving, e.g., the results of data analysis or other complex objects that are difficult to encode into text.
Study its
dump(object, file_handle)andload(file_handle)functions. Then use them to save a dictionary to file, close the Python interpreter, and load the dictionary back from the file.Remark. Make sure that the
file_handleis opened for writing when writing to the file, and for reading when reading from it.
Regular Expressions¶
A regular expression (or regex) is a string that encodes a string *pattern. The pattern specifies which strings do match the regex.
A regex consists of both normal and special characters:
- Normal characters match themselves.
- Special characters match sets of other characters.
A string matches a regex if it matches all of its characters, in the sequence in which they appear.
Example. A few simple examples:
- The regex
"text"matches only the string"text". - The regex
".*"matches all strings. - The regex
"beginning.*"matches all strings that start with"beginning" - The regex
".*end"matches all strings that end with"end".
More formally, the contents of a regex can be are:
| Character | Meaning |
|---|---|
text |
Matches itself |
(regex) |
Matches the regex regex (i.e. parens don’t count) |
^ |
Matches the start of the string |
$ |
Matches the end of the string or just before the newline at the end of the string |
. |
Matches any character except a newline |
regex? |
Matches 0 or 1 repetitions of regex (longest possible) |
regex* |
Matches 0 or more repetitions of regex (longest possible) |
regex+ |
Matches 1 or more repetitions of regex (longest possible) |
regex{m,n} |
Matches from m to n repetitions of regex (longest possible) |
[...] |
Matches a set of characters |
[c1-c2] |
Matches the characters “in between” c1 and c2 |
[^...] |
Matches the complement of a set of characters |
r1|r2 |
Matches both r1 and r2 |
Note
There are many more special symbols that can be used into a regex. We will restrict ourselves to the most common ones.
Example. Let’s start with the anchor characters ^ and $:
- The regex
"^something"matches all strings that start with"something", for instance"something better". - The regex
"worse$"matches all strings that end with"worse", for instance"I am feeling worse".
Example. The “anything goes” character . (the dot) matches all
characters except the newline. For instance:
"."matches all strings that contain at least one character."..."matches all strings that contain at least three characters."^.$"matches all those strings that contain exactly one character."^...$"matches all those strings that contain exactly three characters.
Example. The “optional” character ? match zero or more repetitions of
the preceding regex. For instance:
"words?"matches both"word"and"words": the last character of the"words"regex (that is, the"s") is now optional."(optional)?"matches both"optional"and the empty string."he is (our)? over(lord!)?"matches the following four strings:"he is over","he is our over","he is overlord!", and"he is our overlord!".
Here I used the parens (...) to specify the scope of the ?.
Example. The “zero or more” character "*" and the “one or more” "+"
characters match zero or more (or one or more) repetitions of the preceding
regex. The parens (...) grouping trick still applies. A few examples:
"Python!*"matches all of the following strings:"Python","Python!","Python!!","Python!!!!", etc."(column )+"matches:"column ","column column ", etc. but not the empty string""."I think that (you think that (I think that)*)+ this regex is cool"will match things like"I think that you think that this regex is cool", as well as"I think that you think that I think that you think that I think that this regex is cool", and so on.
Example. The “from n to m” regex {n,m} matches from n to m
repetitions of the previous regex.
For instance, "(column ){2,3}" matches "column column " and "column column column ".
Example. Regexes can also match entire sets of characters (or their complement); in other words, they match all strings containing at least one of the characters in the set. For instance:
"[abc]"matches strings that contain"a","b", or"c". It does not match the string"zzzz"."[^abc]"matches all characters except"a","b", and"c"."[a-z]"matches all lowercase alphabetic characters."[A-Z]"matches all uppercase alphabetic characters."[0-9]"matches all numeric characters from0to9(included)."[2-6]"matches all numeric characters from2to6(included)."[^2-6]"matches all characters except the numeric characters from2to6(included)."[a-zA-Z0-9]"matches all alphanumeric characters.
Example. Perhaps the most powerful special character, the | character,
allows to match either of two regexes. For instance:
"I|you|he|she|it|we|they"will match any string containing at least one of the English personal pronouns."(PRO)+|(SER)+"matches strings like"PRO","PRO PRO","SER"and"SER SER", but not strings like"PRO SER"or"SER PRO SER".
Example. As usual, if you want to “disable” a special character, you have
to escape it, i.e. prefix with a backslash \. An alternative is to insert
the special character to be disabled in square brackets.
For instance, in order to match strings that contain at least one (literal) dot,
you can use either "\." or "[.]".
Example. Regexes can be combined to obtain powerful matching operations. A couple of examples:
A regex that only matches strings that start with
"ATOM", followed by one or more space, followed by three space-separated integers:^ATOM[ ]+[0-9]+ [0-9]+ [0-9]+
The following string matches:
ATOM 30 42 12
A regex that matches a floating-point number in dot-notation:
[0-9]+(\.[0-9]+)?
i.e.
"123"or"2.71828".A regex that matches a floating-point number in mathematical format:
[0-9]+(\.[0-9])?e[0-9]+
i.e.
"6.022e23". (It can be improved!)A regex that matches the following UBR-box sequence motif: zero or more methionins (M), followed by either a glutamic acid (E) or an aspartic acid (D). The motif only appears at the beginning of the sequence, and never at the end (i.e. it is followed by at least one more residue):
^M?([ED]).
The re Module¶
The re module of the Standard Python Library allows to deal with regular
expression matching, for instance checking whether a given string matches a
regular expression, or how many times a regular expression occurs in a string.
The available methods are:
| Returns | Method | Meaning |
|---|---|---|
| match | match(regex, str) | Match a regular expression regex to the beginning of a string |
| match | search(regex, str) | Search a string for the presence of a regex |
| list | findall(regex, str) | Find all occurrences of a regex in a string |
Example. match(regex, string) and search(regex, str) return the
first match of the regex in the string string. The difference is that:
match()requiresregexto match at the beginning ofstringsearch()does not:regexcan match anywhere insidestring
If no match is found, they returns None. Otherwise, a match object is
returned. This makes it easy to see whether the regex matches the string.
To extract the matched text from the match object, call its group()
method, as in the following examples.
For instance (make sure that the re module has been imported):
>>> re.match("nomatch", "some text")
>>> print re.match("nomatch", "some text")
None
searches for the regex "nomatch" into "some text", starting at the
beginning of the target string. Of course, no match is found ("some text"
does not start with "nomatch"), so the function returns None.
Cleary, the following doesn’t work either:
>>> re.match("text", "some text")
>>> print re.match("text", "some text")
None
because "some text" does not start with "text"! However:
>>> print re.search("text", "some text")
<_sre.SRE_Match object at 0x7fac7b5906b0>
>>> print re.search("text", "some text").group()
'text'
works, because search() matches the regex anywhere in string, not
just at the beginning.
In order to see match() work, try:
>>> print re.match("some", "some text")
<_sre.SRE_Match object at 0x7fac7b5906b0>
>>> print re.match("some", "some text").group()
some
Example. findall(regex, text) returns the list of all matches of
regex inside text. For instance, let:
fasta = """>1A3A:A|PDBID|CHAIN|SEQUENCE
MANLFKLGAENIFLGRKAATKEEAIRFAGEQLVKGGYVEPEYVQAMLDREKLTPTYLGESIAVPHGTVEAKDRVLKTGVV
FCQYPEGVRFGEEEDDIARLVIGIAARNNEHIQVITSLTNALDDESVIERLAHTTSVDEVLELLAGRK
>1A3A:B|PDBID|CHAIN|SEQUENCE
MANLFKLGAENIFLGRKAATKEEAIRFAGEQLVKGGYVEPEYVQAMLDREKLTPTYLGESIAVPHGTVEAKDRVLKTGVV
FCQYPEGVRFGEEEDDIARLVIGIAARNNEHIQVITSLTNALDDESVIERLAHTTSVDEVLELLAGRK
>1A3A:C|PDBID|CHAIN|SEQUENCE
MANLFKLGAENIFLGRKAATKEEAIRFAGEQLVKGGYVEPEYVQAMLDREKLTPTYLGESIAVPHGTVEAKDRVLKTGVV
FCQYPEGVRFGEEEDDIARLVIGIAARNNEHIQVITSLTNALDDESVIERLAHTTSVDEVLELLAGRK
>1A3A:D|PDBID|CHAIN|SEQUENCE
MANLFKLGAENIFLGRKAATKEEAIRFAGEQLVKGGYVEPEYVQAMLDREKLTPTYLGESIAVPHGTVEAKDRVLKTGVV
FCQYPEGVRFGEEEDDIARLVIGIAARNNEHIQVITSLTNALDDESVIERLAHTTSVDEVLELLAGRK"""
then we can extract the headers using a single call to findall():
>>> from pprint import pprint
>>> pprint(re.findall("\n>.*\n", fasta))
['\n>1A3A:B|PDBID|CHAIN|SEQUENCE\n',
'\n>1A3A:C|PDBID|CHAIN|SEQUENCE\n',
'\n>1A3A:D|PDBID|CHAIN|SEQUENCE\n']
Warning
Note that the * regex is greedy: it does not stop at the first match of
the | character, but rather greedily proceeds till the last.
To extract only the protein IDs and chain ID, write:
>>> pprint(re.findall("\n>[a-zA-Z0-9:]+[^|]", fasta))
['\n>1A3A:B', '\n>1A3A:C', '\n>1A3A:D']
Exercises - Regular Expressions¶
Note
Here you can find the sequences.fasta FASTA file.
In the following exercises, you can use all the Python tools you want to compute the answer. In other words, looping over the lines of the FASTA file is allowed.
Motifs can appear anywhere in the sequence, unless stated otherwise.
Compute how many sequences contain the following motifs:
- A pheniylalanine (F); two arbitrary amino acids; another phenylalanine. The motif should occur at the end of the sequence.
- An arginine (R); a phenylalanine; an aminoacid that is not a proline (P); an isoleucine (I) or a valine (V).
Compute also how many sequences include at least one of the two motifs.
Compute how many sequences contain the following motifs:
- Three tyrosines (Y); at most three arbitrary amino acids; a histidine (H).
- Contains non-standard or unknown amino acids.
Are there sequences satisfying both conditions?
Note
Standard amino acids: A R N D C E Q G H I L K M F P S T W Y V.
Compute how many sequences contain the following motifs:
- An arginine (R); a lysine (K). The motif should not occur at the beginning of the sequence.
- Two arginines followed by an amino acid that is neither an arginine or a lysine.
- None of the previous two motifs.
Compute how many sequences contain the following motifs:
- A phenylalanyne (F); an arbitrary amino acid; a phenylalanine or a tyrosine (Y); a proline (P).
- A proline; a threonine (T) or a serine (S); an alanine (A); another proline. The motif should be neither at the beginning nor at the end of the sequence.
- The first motif followed by the second, or vice versa.
Biopython¶
What is Biopython?¶
Biopython is the largest and most common bioinformatics package for Python. It contains a number of different sub-modules for common bioinformatics tasks.
It is freely available here:
while the entire source code is hosted here:
Note
The Biopython code is distributed under the Biopython License.
Biopython sports several features. It provides:
Data structures for biological sequences and features thereof, as well as a multitude of manipulation functions for performing common tasks, such as translation, transcription, weight computations, ...
Functions for loading biological data into
dict-like data structures.Supported formats include, for instance: FASTA, PDB, GenBank, Blast, SCOP, PubMed/Medline, ExPASy-related formats
Functions for data analysis, including basic data mining and machine learning algorithm: k-Nearest Neighbors, Naive Bayes, and Support Vector Machines
Access to online services and database, including NCBI services (Blast, Entrez, PubMed) and ExPASY services (SwissProt, Prosite)
Access to local services, including Blast, Clustalw, EMBOSS.
Warning
In order to use local services (such as Blast), the latter must be properly installed and configured on the local machine.
Setting up the local services is beyond the scope of this course.
Note
In order to install Biopython:
On a Debian-based distributions (for instance Ubuntu), just type:
$ sudo apt-get install python-biopython
On a generic GNU/Linux distribution, just type:
$ pip install biopython --user
Please refer to the official documentation for further instructions.
Note
While Biopython is the main player in the field, it is not the only one. Other interesting packages are:
ETE and DendroPy, dedicated to computation and visualization of phylogenetic trees.
CSB for dealing with sequences and structures, computing alignments and profiles (with profile HMMs), and Monte Carlo sampling.
biskit is designed for structural bioinformatics tasks, such as loading 3D protein structures in Protein Data Bank (PDB) format, homology modelling, molecular dynamics simulations (using external simulators) and trajectory computations, and docking, among others.
You are free to mix and match all the packages you need.
Biopython: Sequences¶
Sequences lay at the core of bioinformatics: although DNA, RNA and proteins are molecules with specific structures and dynamical behaviors, their basic building block is their sequence.
Biopython encodes sequences using objects of type Seq, provided by the
Bio.Seq sub-module. Take a look at their manual:
from Bio.Seq import Seq
help(Seq)
Note
The official documentation of the Biopython Seq class can be found on
the Biopython wiki.
Each Seq object is associated to a formal alphabet.
- Each sequence has its own alphabet
- Conversions between different sequence types (transcription, translation) can be seen as a change of alphabet.
- Some operations (e.g. concatenation) can not be done between sequences with incompatible alphabets.
The various alphabets supported by Biopython can be found in Bio.Alphabet,
e.g. Bio.Alphabet.IUPAC.
The most basic alphabets are:
IUPAC.unambiguous_dna: a sequence of DNAIUPAC.unambiguous_rna: a sequence of RNAIUPAC.protein: a protein sequence
Example. To create a generic sequence, i.e. a sequence not bound to any specific alphabet, write:
>>> from Bio.Seq import Seq
>>> s = Seq("ACCGTTTAAC") # no alphabet specified
>>> print type(s)
<class 'Bio.Seq.Seq'>
>>> s
Seq('ACCGTTTAAC', Alphabet())
>>> print s
ACCGTTTAAC
To create a DNA sequence, specify the corresponding alphabet when creating
the Seq object:
>>> from Bio.Seq import Seq
>>> from Bio.Alphabet import IUPAC
>>> s = Seq("ACCGTTTAAC", IUPAC.unambiguous_dna)
>>> s
Seq('ACCGTTTAAC', IUPACUnambiguousDNA())
Seq objects act mostly like standard Python strings str. For instance,
you can write:
>>> from Bio.Seq import Seq
>>> s = Seq("ACTG")
>>> len(s)
4
>>> s[2]
'T'
>>> s[1:-1]
Seq('CT', Alphabet())
>>> s.count("C")
3
However, some operations are restricted to Seq objects with the same
alphabet (in order to prevent the user from, say, concatenating nucleotides
and amino acids):
>>> s = Seq("ACTG", IUPAC.unambiguous_dna)
>>> s + s
Seq('ACTGACTG', IUPACUnambiguousDNA())
>>> t = Seq("AAADGHHHTEWW", IUPAC.protein)
>>> s
Seq('ACTG', IUPACUnambiguousDNA())
>>> t
Seq('AAADGHHHTEWW', IUPACProtein())
>>> s + t
Traceback (most recent call last):
File "<stdin>", line 1, in <module>
File "/home/stefano/.local/lib/python2.7/site-packages/Bio/Seq.py", line 292, in __add__
self.alphabet, other.alphabet))
TypeError: Incompatible alphabets IUPACUnambiguousDNA() and IUPACProtein()
To convert back a Seq object into a normal str object, just use the
normal type-casting operator str():
>>> s = Seq("ACTG", IUPAC.unambiguous_dna)
>>> str(s)
Warning
Just like strings, Seq objects are immutable:
>>> s = Seq("ACCGTTTAAC")
>>> s[0] = "G"
Traceback (most recent call last):
File "<stdin>", line 1, in <module>
TypeError: 'Seq' object does not support item assignment
Thanks to their immutability, Seq objects can be used as keys in
dictionaries (like str objects do).
Note
Biopython also provides a mutable sequence type MutableSeq.
You can convert back and forth between Seq and MutableSeq:
>>> s = Seq("ACTG", IUPAC.unambiguous_dna)
>>> muts = s.tomutable()
>>> muts[0] = "C"
>>> muts
MutableSeq('CCTG', IUPACUnambiguousDNA())
>>> muts.toseq()
Seq('CCTG', IUPACUnambiguousDNA())
Some operations are only available when specific alphabets are used. Let’s see a few examples.
Example. The complement() method allows to complement a DNA or RNA
sequence:
>>> s = Seq("CCGTTAAAAC", IUPAC.unambiguous_dna)
>>> s.complement()
Seq('GGCAATTTTG', IUPACUnambiguousDNA())
while the reverse_complement() also reverses the result from left
to right:
>>> s.reverse_complement()
Seq('GTTTTAACGG', IUPACUnambiguousDNA())
Warning
Neither of these methods is available for other alphabets:
>>> p = Seq("ACLKQ", IUPAC.protein)
>>> p.complement()
Traceback (most recent call last):
File "<stdin>", line 1, in <module>
File "/home/stefano/.local/lib/python2.7/site-packages/Bio/Seq.py", line 760, in complement
raise ValueError("Proteins do not have complements!")
ValueError: Proteins do not have complements!
Example. (Adapted from the Biopython tutorial.) Transcription is the biological process by which a DNA sequence is transcribed into the corresponding DNA.
A picture of the transcription process:
5’ ATGGCCATTGTAATGGGCCGCTGAAAGGGTGCCCGATAG 3’ (coding strand)
|||||||||||||||||||||||||||||||||||||||
3’ TACCGGTAACATTACCCGGCGACTTTCCCACGGGCTATC 5’ (template strand)
|
Transcription
↓
5’ AUGGCCAUUGUAAUGGGCCGCUGAAAGGGUGCCCGAUAG 3’ (mRNA)
The same process can be “simulated” on Seq objects. Starting from the
coding strand (as is usually done in bioinformatics), it is only a matter
of converting T’s into U’s:
>>> coding_dna
Seq('ATGGCCATTGTAATGGGCCGCTGAAAGGGTGCCCGATAG', IUPACUnambiguousDNA())
>>> mrna = coding_dna.transcribe()
>>> mrna
Seq('AUGGCCAUUGUAAUGGGCCGCUGAAAGGGUGCCCGAUAG', IUPACUnambiguousRNA())
From the template strand instead we first have to compute the reverse complement, and then perform the character substitution:
>>> template_dna
'TACCGGTAACATTACCCGGCGACTTTCCCACGGGCTATC'
>>> template_dna.reverse_complement().transcribe()
Seq('AUGGCCAUUGUAAUGGGCCGCUGAAAGGGUGCCCGAUAG', IUPACUnambiguousRNA())
It is also easy to perform the reverse process, back-transcription:
>>> mrna
'AUGGCCAUUGUAAUGGGCCGCUGAAAGGGUGCCCGAUAG'
>>> mrna.back_transcribe()
Seq('ATGGCCATTGTAATGGGCCGCTGAAAGGGTGCCCGATAG', IUPACUnambiguousDNA())
Warning
These functions also take care of checking that the input alphabet is the right one (e.g. DNA) and apply the right alphabet to the result (e.g. RNA).
Example. (Adapted from the Biopython tutorial.) Let’s not take a look at translation, i.e. mRNA to protein.
The code is as follows:
>>>from Bio.Seq import Seq
>>>from Bio.Alphabet import IUPAC
>>>mrna = Seq("AUGGCCAUUGUAAUGGGCCGCUGAAAGGGUGCCCGAUAG", IUPAC.unambiguous_rna)
>>>print mrna
AUGGCCAUUGUAAUGGGCCGCUGAAAGGGUGCCCGAUAG
>>>print mrna.translate()
MAIVMGR*KGAR*
Here the stop codons are indicated with an asterisk "*".
Now, different coding tables are available, for instance:
>>> coding_dna.translate(table="Vertebrate Mitochondrial")
Seq('MAIVMGRWKGAR*', HasStopCodon(IUPACProtein(), '*'))
It’s also possible to tell the translate() method to stop at the first
stop codon:
>>> coding_dna.translate()
Seq('MAIVMGR*KGAR*', HasStopCodon(IUPACProtein(), '*'))
>>> coding_dna.translate(to_stop=True)
Seq('MAIVMGR', IUPACProtein())
In this case the stop codon is not included in the resulting sequence.
Note
The Genetic Codes page of the NCBI provides the full list of translation tables used by Biopython.
In order to visualize the various translation tables, you can do:
from Bio.Data import CodonTable
ttable = CodonTable.unambiguous_dna_by_name["Standard"]
print ttable
ttable = CodonTable.unambiguous_dna_by_name["Vertebrate Mitochondrial"]
print ttable
You can also visualize what are the encodings of stop and start codons:
print ttable.stop_codons
print ttable.start_codons
Let’s take a look at some common sequence formats.
FASTA is the most basic format, we have used it several times already:
>sp|P25730|FMS1_ECOLI CS1 fimbrial subunit A precursor (CS1 pilin) MKLKKTIGAMALATLFATMGASAVEKTISVTASVDPTVDLLQSDGSALPNSVALTYSPAV NNFEAHTINTVVHTNDSDKGVVVKLSADPVLSNVLNPTLQIPVSVNFAGKPLSTTGITID SNDLNFASSGVNKVSSTQKLSIHADATRVTGGALTAGQYQGLVSIILTKST >sp|P15488|FMS3_ECOLI CS3 fimbrial subunit A precursor (CS3 pilin) MLKIKYLLIGLSLSAMSSYSLAAAGPTLTKELALNVLSPAALDATWAPQDNLTLSNTGVS NTLVGVLTLSNTSIDTVSIASTNVSDTSKNGTVTFAHETNNSASFATTISTDNANITLDK NAGNTIVKTTNGSQLPTNLPLKFITTEGNEHLVSGNYRANITITSTIK
GenBank is a richer sequence format for genes, it includes fields for various kinds of annotations (e.g. source organism, bibliographical references, database-specific accession number).
A GenBank file looks like this:
LOCUS 5I3U_F 39 bp DNA linear SYN 31-AUG-2016 DEFINITION Chain F, Structure Of Hiv-1 Reverse Transcriptase N-site Complex; Catalytic Incorporation Of Aztmp To A Dna Aptamer In Crystal. ACCESSION 5I3U_F VERSION 5I3U_F KEYWORDS . SOURCE synthetic construct ORGANISM synthetic construct other sequences; artificial sequences. REFERENCE 1 (bases 1 to 39) AUTHORS Tu,X., Das,K., Han,Q., Bauman,J.D., Clark AD,Jr., Hou,X., Frenkel,Y.V., Gaffney,B.L., Jones,R.A., Boyer,P.L., Hughes,S.H., Sarafianos,S.G. and Arnold,E. TITLE Structural basis of HIV-1 resistance to AZT by excision JOURNAL Nat. Struct. Mol. Biol. 17 (10), 1202-1209 (2010) PUBMED 20852643 ... FEATURES Location/Qualifiers source 1..39 /organism="synthetic construct" /mol_type="other DNA" /db_xref="taxon:32630" ORIGIN 1 taatacncnc ccttcggtgc tttgcaccga agggggggn //
UniProt (obtained by combining SwissProt, TrEMBL and PIR) is the GenBank equivalent for protein sequences:
ID 143B_HUMAN STANDARD; PRT; 245 AA. AC P31946; DT 01-JUL-1993 (Rel. 26, Created) DT 01-FEB-1996 (Rel. 33, Last sequence update) DT 15-SEP-2003 (Rel. 42, Last annotation update) DE 14-3-3 protein beta/alpha (Protein kinase C inhibitor protein-1) DE (KCIP-1) (Protein 1054). GN YWHAB. OS Homo sapiens (Human). OC Eukaryota; Metazoa; Chordata; Craniata; Vertebrata; Euteleostomi; OC Mammalia; Eutheria; Primates; Catarrhini; Hominidae; Homo. OX NCBI_TaxID=9606; RN [1] RP SEQUENCE FROM N.A. RC TISSUE=Keratinocytes; RX MEDLINE=93294871; PubMed=8515476; RA Leffers H., Madsen P., Rasmussen H.H., Honore B., Andersen A.H., RA Walbum E., Vandekerckhove J., Celis J.E.; RT "Molecular cloning and expression of the transformation sensitive RT epithelial marker stratifin. A member of a protein family that has RT been involved in the protein kinase C signalling pathway.";
The Bio.SeqIO module provides a functions to parse all of the above
(among others).
In particular, the parse() function takes a file handle and the format
of the file (as a string) and returns an iterable of SeqRecord objects:
from Bio.SeqIO import parse
help(parse)
f = open("test.fasta")
for record in parse(f, "fasta"):
print record.id, record.seq
Warning
An iterable is a function that allows to iterate over a collection of objects (similar to a list).
When given an iterator, you can either use it as in the code above:
iterable = get_it_from_somewhere() # e.g. parse()
for element in iterable:
do_something(element)
or extract one element at a time, with the next() method:
iterable = get_it_from_somewhere() # e.g. parse()
first_element = iterable.next()
do_something(first_element)
second_element = iterable.next()
do_something(second_element)
# ...
The iterable allows to iterate over a list of SeqRecord objects. The
SeqRecord objects associate a Seq sequence with additional meta-data,
such as human-readable descriptions, annotations, and sequence features:
>>> from Bio.SeqRecord import SeqRecord
>>> help(SeqRecord)
Each SeqRecord object provides these attributes:
.seq: the sequence itself, typically aSeqobject..id: the primary identifier of the sequence – a string..name: a “common” identifier for the sequence, also a string..description: a human readable description of the sequence..annotations: adictof additional information.
among others.
In order to read a SeqRecord object from a file:
>>> from Bio import SeqIO
>>> record = SeqIO.parse(open("sequences.fasta"), "fasta").next()
>>> record
SeqRecord(seq=Seq('METNCRKLVSACVQLGVQPAAVECLFSKDSEIKKVEFTDSPESRKEAASSKFFP...RPV', SingleLetterAlphabet()), ...
>>> record.id
'Q99697'
>>> record.name
'Q99697'
>>> record.annotations
{}
Here record.annotations is empty because the FASTA format does not really
support sequence annotations.
The very nice thing about the SeqIO and SeqRecord mechanism is that
they allow to process all of the different formats described above (FASTA,
GenBank, UniProt) using the very same code!
Let’s see what happens with a GenBank file:
>>> record = SeqIO.parse(open("data.genbank"), "genbank").next()
>>> record
SeqRecord(seq=Seq('TAATACNCNCCCTTCGGTGCTTTGCACCGAAGGGGGGGN', IUPACAmbiguousDNA()), ...
>>> from pprint import pprint
>>> pprint(record.annotations)
{'accessions': ['5I3U_F'],
'data_file_division': 'SYN',
'date': '31-AUG-2016',
'keywords': [''],
'organism': 'synthetic construct',
'references': [Reference(title='Structural basis of HIV-1 resistance to AZT by excision', ...)],
'source': 'synthetic construct',
'taxonomy': ['other sequences', 'artificial sequences'],
'topology': 'linear'}
This way, you can (almost) forget about the format of the sequence data you are working with – all the parsing is done automatically by Biopython.
Finally, the SeqIO.write() method allows to write a SeqRecord objects
or a list of SeqRecord objects to disk, in a given format:
fr = open("data.genbank", "r")
record = SeqIO.parse(fr, "genbank").next()
fw = open("seq1.fasta", "w)
SeqIO.write(record, fw, "fasta")
Biopython: Structures¶
Biopython allows to manipulate polypeptide structures via the Bio.PDB module.
We will focus on data coming from the Protein Data Bank (PDB):
The PDB is by far the largest protein structure resource available online. At the time of writing, it hosts more thank 120k distinct protein structures, including protein-protein, protein-DNA, protein-RNA complexes.
In order to load the required code, just type:
from Bio.PDB import *
Note
For the documentation of the Bio.PDB module, see:
Note
In the following we will use the Zaire Ebolavirus glicoprotein structure
5JQ3, available here:
This is what this structure looks like:
Protein Structure File Formats¶
The PDB distributes protein structures in two different formats:
The XML-based file format. It is not supported by Biopython.
The
mmciffile format. (The actual file extension is.cif.) See for instance:This is the most recent and “parsable” file format.
The
pdbfile format, which is a specially formatted text file. See for instance:This is the historical file format. It is (mostly) still distributed because of legacy tools that do not (yet) understand the newer and cleaner
mmciffiles.
Warning
Some (many?) PDB files distributed by the Protein Data Bank contain formatting errors that make them ambiguous or difficult to parse.
The Bio.PDB module attempts to deal with these errors automatically.
Of course, Biopython is not perfect, and some formatting errors may still make it do the wrong thing, or raise an exception.
Depending on your goal, you can either:
- manually correct the structure, e.g. if you have to analyze a single structure
- ignore the error, e.g. when you are just computing global statistics about a large number of structures, and a few errors do not really impact the overall numbers
We will see how to ignore these “spurious” errors soon.
The Bio.PDB module implements two different parsers, one for the pdb
format and one for the mmcif format. Let’s try them out.
Starting with:
>>> from Bio.PDB import *
To load a cif file, use the Bio.MMCIF.MMCIFParser module:
>>> parser = MMCIFParser()
>>> struct = parser.get_structure("5jq3", "5jq3.cif")
# four warnings
>>> struct
<Structure id=5jq3>
>>> print type(struct)
<class 'Bio.PDB.Structure.Structure'>
To load a pdb file, use the Bio.PDB.PDBParser module:
>>> parser = PDBParser()
>>> struct = parser.get_structure("5jq3", "5jq3.pdb")
# four warnings
>>> struct
<Structure id=5jq3>
>>> print type(struct)
<class 'Bio.PDB.Structure.Structure'>
As you can see, both parsers give you the same kind of object, a Structure.
The header of the structure is a dict which stores meta-data about the
protein structure, including its resolution (in Angstroms), name, author,
release date, etc.
To access it, write:
>>> print struct.header.keys()
['structure_method', 'head', 'journal', 'journal_reference',
'compound', 'keywords', 'name', 'author', 'deposition_date',
'release_date', 'source', 'resolution', 'structure_reference']
>>> print struct.header["name"]
crystal structure of ebola glycoprotein
>>> print struct.header["release_date"]
2016-06-29
>>> print struct.header["resolution"]
2.23 # Å
Warning
The header dictionary may be an empty, depending on the structure and
the parser. Try it out with the mmcif parser!
Note
You can download the required files directly from the PBD server with:
>>> pdbl = PDBList()
>>> pdbl.retrieve_pdb_file('1FAT')
Downloading PDB structure '1FAT'...
'/home/stefano/scratch/fa/pdb1fat.ent'
The file will be put into the current working directory.
The PDBList object also allows you to retrieve a full copy of the PDB
database, if you wish to do so. Note that the database takes quite a bit of
disk space.
Structures as Hierarchies¶
Structural information is natively hierarchical.
The hierarchy is represented by Bio.PDB as follows (image taken from the
Biopython Structural Bioinformatics FAQ):
Example. For instance, the 5JQ3 PDB structure is composed of:
- A single model, containing
- Two chains:
- chain A, containing 338 residues
- chain B, containing 186 residues
- Each residue is composed by multiple atoms, each having a 3D position represented as by (x, y, z) coordinates.
- Two chains:
More specifically, here are the related Bio.PDB classes:
A
Structuredescribes one or more models, i.e. different 3D conformations of the very same structure (for NMR).The
Structure.get_models()method returns an iterator over the models:>>> it = struct.get_models() >>> it <generator object get_models at 0x7f318f0b3fa0> >>> models = list(it) >>> models [<Model id=0>] >>> type(models[0]) <class 'Bio.PDB.Model.Model'>
Here I turned the iterator into a proper list with the
list()function.A
Structurealso acts as adict: given a model ID, it returns the correspondingModelobject:>>> struct[0] <Model id=0>
A
Modeldescribes exactly one 3D conformation. It contains one or more chains.The
Model.get_chain()method returns an iterator over the chains:>>> chains = list(models[0].get_chains()) >>> chains [<Chain id=A>, <Chain id=B>] >>> type(chains[0]) <class 'Bio.PDB.Chain.Chain'> >>> type(chains[1]) <class 'Bio.PDB.Chain.Chain'>
A
Modelalso acts as adictmapping from chain IDs toChainobjects:>>> model = models[0] >>> model <Model id=0> >>> model["B"] <Chain id=B>
A
Chaindescribes a proper polypeptide structure, i.e. a consecutive sequence of bound residues.The
Chain.get_residues()method returns an iterator over the residues:>>> residues = list(chains[0].get_residues()) >>> len(residues) 338 >>> type(residues[0]) <class 'Bio.PDB.Residue.Residue'>
A
Residueholds the atoms that belong to an amino acid.The
Residue.get_atom()(unfortunately, “atom” is singular) returns an iterator over the atoms:>>> atoms = list(residues[0].get_atom()) >>> atoms [<Atom N>, <Atom CA>, <Atom C>, <Atom O>, <Atom CB>, <Atom OG>] >>> type(atoms[0]) <class 'Bio.PDB.Atom.Atom'>
An
Atomholds the 3D coordinate of an atom, as aVector:>>> atoms[0].get_vector() <Vector -68.82, 17.89, -5.51>
The x, y, z coordinates in a
Vectorcan be accessed with:>>> vector = atoms[0].get_vector() <Vector -68.82, 17.89, -5.51> >>> coords = list(vector.get_array()) [-68.81500244, 17.88899994, -5.51300001]
The other methods of the
Vectorclass allow to compute theangle()with another vector, compute itsnorm(), map it to the unit sphere withnormalize(), and multiply the coordinates by a matrix withleft_multiply().
Example. To iterate over all atoms in a structure, type:
parser = PDBParser()
struct = parser.get_structure("target", "5jq3.pdb")
for model in struct:
for chain in model:
for res in chain:
for atom in res:
print atom
and to print a single atom:
>>> struct[0]["B"][502]["CA"].get_vector()
<Vector -64.45, 8.02, 1.62>
There are also shortcuts to skip the middle levels:
for res in model.get_residues():
print residue
and you can go back “up” the hierarchy:
model = chain.get_parent()
Finally, you can inspect the properties of atoms and residues using the methods described here:
The meaning of residue IDs are not obvious. From the Biopython Structural Bioinformatics FAQ:
What is a residue id?
This is a bit more complicated, due to the clumsy PDB format. A residue id is a tuple with three elements:
- The hetero-flag: this is
"H_"plus the name of the hetero-residue (e.g. ‘H_GLC’ in the case of a glucose molecule), or ‘W’ in the case of a water molecule.- The sequence identifier in the chain, e.g. 100
- The insertion code, e.g. ‘A’. The insertion code is sometimes used to preserve a certain desirable residue numbering scheme. A Ser 80 insertion mutant (inserted e.g. between a Thr 80 and an Asn 81 residue) could e.g. have sequence identifiers and insertion codes as follows: Thr 80 A, Ser 80 B, Asn 81. In this way the residue numbering scheme stays in tune with that of the wild type structure. The id of the above glucose residue would thus be (‘H_GLC’, 100, ‘A’). If the hetero-flag and insertion code are blank, the sequence identifier alone can be used:
# Full id residue = chain[(' ', 100, ' ')] # Shortcut id residue = chain[100]The reason for the hetero-flag is that many, many PDB files use the same sequence identifier for an amino acid and a hetero-residue or a water, which would create obvious problems if the hetero-flag was not used.
Writing a pdb file¶
Given a Structure object, it is easy to write it to disk in pdb format:
struct = ... # a Structure
io = PDBIO()
io.set_structure(struct)
io.save("output.pdb")
And that’s it!
Structure Alignment¶
In order to be comparable, structures must first be aligned: superimposed on top of each other.
Note
Structure alignment is very common problem.
Say you have a wildtype protein and an SNP mutant, and you would like to compare the structural consequences of the mutation. Then in order to compare the structure, first you want them to match “as closely as possible”.
It also occurs in protein structure prediction: in order to evaluate the quality of a structural predictor (a software that takes the sequence of a protein and attempts to figure out how the structure looks like), the predictions produced by the software must be compared to the real corresponding protein structures. The same goes, of course, for dynamical molecular simulation software.
For a quick overview of several superposition methods, see:
The most basic superposition algorithms work by rototranslating the two proteins so to maximize their alignment.
In particular, the quality of the alignment is measured by the Root Mean Squared Error (RMSD), defined as:
This is a least-squares minimization problem, and is solved using standard decomposition techniques.
Warning
This is a useful, but naive interpretation of the structural alignment problem. It may not be suitable to the specific task.
For instance, it is well known that there is a correlation between the RMSD values and the length of the protein, which makes average RMSD values computed over different proteins very biased.
Different models of structure alignment avoid this issue, such as TM-score:
We will not cover them here.
The Bio.PDB module supports RMSD-based structural alignment via the
Superimposer class, let’s see how.
First, let’s extract the list of atoms from the two chains of our structure:
>>> atoms_a = list(struct[0]["A"].get_atoms())
>>> atoms_b = list(struct[0]["B"].get_atoms())
We make sure that the two lists of atoms are of the same length; the
Superimposer does not work otherwise:
>>> atoms_a = atoms_a[:len(atoms_b)]
Now we create a Superimposer object and specify the two lists of atoms that
should be aligned:
>>> si = Superimposer()
>>> si.set_atoms(atoms_a, atoms_b)
Here the atoms atoms_a are fixed, while the atoms atoms_b are movable,
i.e. the second structure is aligned on top of the first one.
The superposition occurs automatically, behind the scene. We can access
the minimum RMSD value from the si object with:
>>> print si.rms
26.463458137
as well as the rotation matrix and the translation vector:
>>> rotmatrix = si.rotran[0]
>>> rotmatrix
array([[-0.43465167, 0.59888164, 0.67262078],
[ 0.63784155, 0.73196744, -0.23954504],
[-0.63579563, 0.32490683, -0.70014246]])
>>> transvector = si.rotran[1]
>>> transvector
Finally, we can apply the optimal rototranslation to the movable atoms:
array([-92.66668863, 31.1358793 , 17.84958153])
>>> si.apply(atoms_b)
Biopython (Exercises)¶
Biopython: Sequences¶
Note
The sequences.fasta file is available at:
Write a Python program that asks the user for two DNA sequences, and prints the reverse complement of their concatenation.
Write a Python program that asks the user for a DNA sequence, and prints both the corresponding mRNA sequence and protein sequence, including stop codons (according to the standard translation table).
Write a Python program that asks the user for a DNA sequence, and prints both the corresponding mRNA sequence and protein sequence, including stop codons (according to the Yeast Mitochondrial Code translation table).
Write a Python program that takes the sequence of the 1AI4 PDB protein (download the FASTA file manually), and writes a corresponding UniProt file.
Write a Python program that takes the
sequences.fastafile and writes N single-sequence FASTA files, calledsequence{number}.fasta, each one containing a single sequence of the original file.Do the same, but create N GenBank files instead.
Write a Python program that takes the
sequences.fastafile and writes arevcomp.fastafile with the reverse complements of the original sequences.Hint. The
SeqIO.write()function can write an entire list ofSeqIOrecordsSolve Exercise 3 of the Programs section using Biopython where appropriate.
Solve Exercise 2 of the Programs section using Biopython where appropriate.
Hint. Study carefully the
.annotationsof theSeqRecordobtained by parsing the UniProt file.Find and download a single sequence record from genbank. The genbank identifier of the record is HE805982.1. This record contains information about the DNA region coding for HBx, a multifunctional hepatitis B viral protein involved in modulating several pathways by directly or indirectly interacting with hosts factors (protein degradation, apoptosis, transcription, signal transduction, cell cycle progress, and genetic stability). Write a Python program that, using the genbank record, saves the corresponding protein sequence in fasta format.
From UniProt find and download the records relative to the four human ELAV proteins (ELAVL1: Q15717, ELAVL2: Q12926, ELAVL3: Q14576, ELAVL4: P26378). Download each record in text format and store the four records in a dedicated directory. Now write a python program that takes all the uniprot files in the directory and appends all the sequences in fasta format, to the file created in the previous exercise. Can you print sequences ordered by increasing length?
Hint. The
os.listdir()function in theosmodule can save in a list all the names of the files in a directory.Write a program that, given a fasta file containing multiple protein sequences and a string specified by the user, prints to a new file only sequences that contain at least one occurrence of the string (regular expressions are allowed). Test your program with the file
sequences.fasta, printing sequences containing a stretch of at least three glutamines.Try to execute exercises 20.1.6 and 20.1.7 in the biopython tutorial page:
Biopython: Structures¶
Note
The P53 files (FASTA and three PDB files) are available here:
Ask the user for the path to a
pdbfile. Print the header of the PDB file on screen, including the name, author, resolution and release date of the PDB structure.Same as above, but for an
mmciffile.Ask the user for the path to a
pdbfile. Print the number of models it contains. For each model, print the name length of its chains.Same as above, but for an
mmciffile.Ask the user for the path to a
pdbfile, as well as the name of a chain. Print the number of atoms that each of its residues has.Ask the user for the path to a
pdbfile, and convert its sequence to aFASTAfile.Note. You will need to convert from three-letter amino acid codes to one-letter codes.
Hint. You can use the
SeqIOmodule, but it is not mandatory.Ask the user for the ID of a PDB structure (e.g.
"1A3A"), and download it somewhere using thePDBListclass. Then print the hierarchy of the structure: how many models; for each model how many chains; for each chain how many residues.Note. Try to make the output look pretty.
Ask the user the path to two
pdbfiles, then perform structural alignment on them.Ask the user the path to two
pdbfiles and afloatthreshold. For each chain in the first structure, compare it to each chain in the second structure: if any two residues in the first chain is close enough to a residue of the second chain (i.e. their distance is smaller than the threshold), print the chain ID and residue type of the two residues, as well as their distance.Given the files
1TSR.pdbandwt_1tsr_B.pdbin the P53 package, make sure that they have exactly the same residues.Given the files
wt_1tsr_B.pdbandwt_1tsr_B_nowater.pdbin the P53 package, make sure that the only missing molecules are the"HOH"ones.Hint. Use the
heteroattribute of the residues.
Biopython (Solutions)¶
Pandas¶
What is Pandas?¶
Pandas is a freely available library for loading, manipulating, and visualizing sequential and tabular data, for instance time series or microarrays.
Its main features are:
- Loading and saving with “standard” tabular files, like CSV (Comma-separated Values), TSV (Tab-separated Values), Excel files, and database formats
- Flexible indexing and aggregation of series and tables
- Efficient numerical and statistical operations (e.g. broadcasting)
- Pretty, straightforward visualization
This list is far from complete!
Documentation & Source¶
A few websites worth visiting are the official Pandas website:
as well as the official documentation:
The Pandas source code, available here:
is distributed under the BSD license:
Importing Pandas¶
In order to import Pandas, the customary idiom is:
>>> import pandas as pd
>>> help(pd)
but feel free to use this instead:
>>> from pandas import *
To load the (optional, but super-useful) visualization library, write:
>>> import matplotlib.pyplot as plt
For now, the only method you need is the plt.show() method.
Pandas: A short demonstration¶
Let’s start with a copy of Fisher’s own iris dataset:
The dataset records petal length/width and sepal length/width of three different kinds of iris species: Iris setosa, Iris versicolor, and Iris virginica.
Note
The iris dataset has quite some history – there is even a Wikipedia page about it!:
This is what the iris dataset in CSV format looks like:
SepalLength,SepalWidth,PetalLength,PetalWidth,Name
5.1,3.5,1.4,0.2,Iris-setosa
4.9,3.0,1.4,0.2,Iris-setosa
...
5.0,3.3,1.4,0.2,Iris-setosa
7.0,3.2,4.7,1.4,Iris-versicolor
6.4,3.2,4.5,1.5,Iris-versicolor
...
5.7,2.8,4.1,1.3,Iris-versicolor
6.3,3.3,6.0,2.5,Iris-virginica
5.8,2.7,5.1,1.9,Iris-virginica
...
5.9,3.0,5.1,1.8,Iris-virginica
In an effort to understand the dataset, we would like to visualize the relation between the four properties for the case of Iris virginica.
In pure Python, we would have to:
- Load the dataset by parsing all the rows in the file, then
- Keep only the rows pertaining to Iris virginica, then
- Compute statistics on the values of the rows, making sure to convert from
strings to
float‘s as required, and then - Actually draw the plots by using a specialized plotting library.
With Pandas, this all becomes very easy. Check this out:
>>> import pandas as pd
>>> from pandas.tools.plotting import scatter_matrix
>>> import matplotlib.pyplt as plt
>>> df = pd.read_csv("iris.csv")
>>> scatter_matrix(df[df.Name == "Iris-virginica"])
>>> plt.show()
and that’s it! The result is:
Attention
It only looks bad because I used the default colors. Plots can be customized to your liking!
Of course, there is a lot more to Pandas than solving simple tasks like this.
Pandas datatypes¶
Pandas provides a couple of very useful datatypes, Series and DataFrame:
Seriesrepresents 1D data, like time series, calendars, the output of one-variable functions, etc.DataFramerepresents 2D data, like a column-separated-values (CSV) file, a microarray, a database table, a matrix, etc.
A DataFrame is composed of multiple Series. More specifically, each
column of a DataFrame is a Series. That’s why we will see how the
Series datatype works first.
Attention
Most of what we will say about Series also applies to DataFrame‘s.
The similarities include how indexing is done, how broadcasting applies
to arithmetical operators and Boolean masks, how to compute statistics,
how to deal with missing values (nan‘s) and how plotting is done.
Pandas: Series¶
A Series is a one-dimensional array with a labeled axis. It can hold
arbitrary objects. It works a little like a list and a little like a
dict.
The axis is called the index, and can be used to access the elements; it is
very flexible, and not necessarily numerical.
To access the online documentation, type:
>>> s = pd.Series()
>>> help(s)
Creating a Series¶
Use the Series() constructor to create a new Series object, as follows.
When creating a Series, you can either specify both the date and the
index, like this:
>>> s = pd.Series(["a", "b", "c"], index=[2, 5, 8])
>>> s
2 a
5 b
8 c
dtype: object
>>> s.index
Int64Index([2, 5, 8], dtype='int64')
or pass a single dict in, in which case the index is built from the
dictionary keys:
>>> s = pd.Series({"a": "A", "b": "B", "c": "C"})
>>> s
a A
b B
c C
dtype: object
>>> s.index
Index([u'a', u'b', u'c'], dtype='object')
or skip the index altogether: in this case, the default index (i.e. a
numerical index starting from 0) is automatically assigned:
>>> s = pd.Series(["a", "b", "c"])
>>> s
0 a
1 b
2 c
dtype: object
>>> s.index
RangeIndex(start=0, stop=3, step=1)
Finally, if given a single scalar (e.g. an int), the Series constructor
will replicate it for all indices:
>>> s = pd.Series(3, index=range(10))
>>> s
0 3
1 3
2 3
3 3
4 3
5 3
6 3
7 3
8 3
9 3
dtype: int64
Note
The type of the index changes in the previous exercises.
The library uses different types of index for performance reasons. The
Series class however works (more or less) the same in all cases, so
you can ignore these differences in practical applications.
Accessing a Series¶
Let’s create a Series representing the hours of sleep we had the chance to
get each day of the past week:
>>> sleephours = [6, 2, 8, 5, 9]
>>> days = ["mon", "tue", "wed", "thu", "fri"]
>>> s = pd.Series(sleephours, index=days)
>>> s
mon 6
tue 2
wed 8
thu 5
fri 9
dtype: int64
Now we can access the various elements of the Series both by their label
(i.e. the index) like we would with a dict:
>>> s["mon"]
6
as well as by their position, like we would with a list:
>>> s[0]
6
Warning
If the label or position is wrong, you get either a KeyError:
>>> s["sat"]
KeyError: 'sat'
or an IndexError:
>>> s[9]
IndexError: index out of bounds
We can also slice the positions, like we would with a list:
>>> s[-3:]
wed 8
thu 5
fri 9
dtype: int64
Note that both the data and the index are extracted correctly. It also works with labels:
>>> s["tue":"thu"]
tue 2
wed 8
thu 5
dtype: int64
The first and last n elements can be extracted also using head() and
tail():
>>> s.head(2)
mon 6
tue 2
dtype: int64
>>> s.tail(3)
wed 8
thu 5
fri 9
dtype: int64
Most importantly, can also explicitly pass in a list of positions, like this:
>>> s[[0, 1, 2]]
mon 6
tue 2
wed 8
dtype: int64
>>> s[["mon", "wed", "fri"]]
mon 6
wed 8
fri 9
dtype: int64
>>> s.head()
Warning
Passing in a tuple of positions does not work!
Operator Broadcasting¶
The Series class automatically broadcasts arithmetical operations by a
scalar to all of the elements. Consider adding 1 to our Series:
>>> s
mon 6
tue 2
wed 8
thu 5
fri 9
dtype: int64
>>> s + 1
mon 7
tue 3
wed 9
thu 6
fri 10
dtype: int64
As you can see, the addition was applied to all the entries! This also applies to multiplication and other arithmetical operations:
>>> s * 2
mon 12
tue 4
wed 16
thu 10
fri 18
dtype: int64
This is extremely useful: it frees the analyst from having to write loops. The (worse) alternative the above would be:
for x in s.index:
s[x] += 1
which is definitely more verbose and error-prone, and also takes more time to execute!
Note
The concept of operator broadcasting was taken from the numpy
library, and is one of the key features for writing efficient,
clean numerical code in Python.
In a way, it is a “generalized” version of scalar products (from linear algebra).
The rules govering how broadcasting is applied can be pretty complex (and confusing). Here we will cover constant broadcasting only.
Masks and Filtering¶
Most importantly, broadcasting also applies to conditions:
>>> s > 6
mon True
tue False
wed True
thu False
fri True
dtype: bool
Here the result is a Series with the same index as the original (that is,
the very same labels), and each label label is associated to the result of
the comparison s[label] > 6. This kind of series is called a mask.
Is it useful in any way?
Yes, of course! Masks can be used to filter the elements of a Series
according to a given condition:
>>> s[s > 6]
mon 7
wed 9
fri 10
dtype: int64
Here only those entries corresponding to True‘s in the mask have been
kept. For extracting the elements that satisfy the opposite of the condition,
write:
>>> s[s <= 6]
tue 3
thu 6
dtype: int64
or:
>>> s[~(s > 6)]
tue 3
thu 6
dtype: int64
The latter version is useful when the condition you are checking for is complex and can not be inverted easily.
Automatic Label Alignment¶
Operations between multiple time series are automatically aligned by label, meaning that elements with the same label are matched prior to carrying out the operation.
Consider this example:
>>> s[1:] + s[:-1]
fri NaN # <-- not common
mon NaN # <-- not common
thu 10.0 # <-- common element
tue 4.0 # <-- common element
wed 16.0 # <-- common element
dtype: float64
As you can see, the elements are summed in the right order! But what’s up with
those nan‘s?
The index of the resulting Series is the union of the indices of
the operands. What happens depend on whether a given label appears in both
ipnut Series or not:
- For common labels (in our case
"tue","wed","thu"), the outputSeriescontains the sum of the aligned elements. - For labels appearing in only one of the operands (
"mon"and"fri"), the result is anan, i.e. not-a-number.
Warning
nan means not a number.
It is just a symbolic constant that specifies that the object is a number-like entity with an invalid or undefined value. Think for instance of the result of division by 0.
In the example above, the result is a nan because it represents the
sum of an element, like s[1:]["fri"], and a non-existing element,
like s[:-1]["fro"].
It is used to indicate undefined results (like above) or missing measurements (as we will see).
The actual value of the nan symbol is taken from numpy, see
numpy.nan. You can actually use it to insert nan‘s manually:
>>> s["fri"] = np.nan
>>> s
mon 6.0
tue 2.0
wed 8.0
thu 5.0
fri NaN
dtype: float64
Dealing with Missing Data¶
By default, missing entries and invalid results in Pandas are defined as
nan. Consider the Series from the previous example:
>>> t = s[1:] + s[:-1]
>>> t
fri NaN
mon NaN
thu 12.0
tue 6.0
wed 18.0
dtype: float64
There are different strategies for dealing with nan‘s. There is no “best”
strategy: you have to pick one depending on the problem you are trying to
solve.
Drop all entries that have a
nan, withdropna()(read it as drop N/A, i.e. not available):>>> nonan_t = t.dropna() >>> nonan_t thu 12.0 tue 6.0 wed 18.0 dtype: float64
Filling in the
nanentries with a more palatable value, for instance a default value that does not disrupt your statistics:>>> t.fillna(0.0) fri 0.0 mon 0.0 thu 12.0 tue 6.0 wed 18.0 dtype: float64
or:
>>> t.fillna("unknown") fri unknown mon unknown thu 12 tue 6 wed 18 dtype: object
Filing in the
nanby imputing the missing value from the surrouding ones.>>> s mon NaN tue 2.0 wed 8.0 thu NaN fri NaN dtype: float64 >>> s.ffill() mon NaN tue 2.0 wed 8.0 thu 8.0 fri 8.0 dtype: float64
Warning
Most of Pandas function gracefully deal with missing values.
For instance the mean() method has a skipna argument can be either
True or False. If it is True, the mean will not consider the
missing values; if it is False, the result of a series containing
a nan will itself be a nan (as should happen in purely mathematical
terms).
Computing Statistics¶
Series objects support a variety of mathematical and statistical
operations:
The sum and product of all values in the
Series>>> s.sum() 30 >>> s.prod() 11340
The cumulative sum:
>>> s.cumsum() mon 6 tue 8 wed 16 thu 21 fri 30 dtype: int64
The minimum and maximum values and their indices:
>>> s.max() 9 >>> s.argmax() 'fri' >>> s[s.argmax()] 9
and the same for
s.min()ands.argmin().The mean, variance, std. deviation, median, quantiles, etc.:
>>> s.mean() 6.0 >>> s.var() 7.5 >>> s.std() 2.7386127875258306 >>> s.median() 6.0 >>> s.quantile([0.25, 0.5, 0.75]) 0.25 6.0 0.50 7.0 0.75 9.0 dtype: float64
These can be paired with masks for awesome extraction effects:
>>> s[s > s.quantile(0.66)] wed 9 fri 10 dtype: int64
The Pearson, Kendall, Spearman correlations between series:
>>> s.corr(s) # Pearson by default 1.0 >>> s.corr(s, method="spearman") 0.99999999999999989
The autocorrelation with arbitrary time lag:
>>> s.autocorr(lag=0) 1.0 >>> s.autocorr(lag=1) -0.54812812776251907 >>> s.autocorr(lag=2) 0.99587059488582252
... and many, many others.
Note
A quick way to examine a Series is to use the describe() method:
>>> s.describe()
count 5.000000
mean 7.000000
std 2.738613
min 3.000000
25% 6.000000
50% 7.000000
75% 9.000000
max 10.000000
dtype: float64
Warning
You are not required to remember all these methods!
Just be aware that if you need basic statistical tool, probably pandas
has them.
Plotting¶
We want to visualize our simple sleeping hours data:
>>> sleephours = [6, 2, 8, 5, 9]
>>> days = ["mon", "tue", "wed", "thu", "fri"]
>>> s = pd.Series(sleephours, index=days)
Now that we have a Series, let’s import the additional matplotlib
module:
>>> import matplotlib.pyplot as plt
This is the customary idiom to import the library. Let’s plot the data!
With a simple line plot:
>>> s.plot() >>> plt.show()
With a more readable bar plot:
>>> s.plot(kind="bar") >>> plt.show()
With a histogram:
>>> s.plot(kind="hist") >>> plt.show()
Note
There are very many ways to improve the looks of your plots, like changing the shape, size, margins, colors, labels, etc.
Browse the documentation for inspiration:
Note
To save the plot to a file instead of merely showing it on screen, use the
savefig() method of the matplotlib library:
>>> plt.savefig("output.png")
Pandas: DataFrame¶
Pandas DataFrame is the 2D analogue of a Series: it is essentially a
table of heterogeneous objects.
Recall that a Series holds both the values and the labels of all its
elements. Analogously, a DataFrame two major attributes:
- the
index, which holds the labels of the rows - the
columns, which holds the labels of the columns - the
shape, which describes the dimension of the table
Each column in the DataFrame is a Series: when you extract a column
from a DataFrame you get a proper Series, and you can operate on it
using all the tools presented in the previous section.
Further, most of the operations that you can do on a Series, you can also
do on an entire DataFrame. The sample principles apply: broadcasting, label
alignment, and plotting. They all work very similarly.
Warning
Not all operations work exactly the same on the two datatypes.
This is of course because some operations that make sense on a 2D table, do not really apply to a 1D sequence, and vice versa.
However the analogy holds well enough in most cases.
Creating a DataFrame¶
Just like for Series, there are various options. We can:
Create a
DataFramefrom a dictionary ofSeries:>>> d = { ... "name": ... pd.Series(["bobby", "ronald", "ronald", "ronald"]), ... "surname": ... pd.Series(["fisher", "fisher", "reagan", "mcdonald"]), ... } >>> df = pd.DataFrame(d) >>> df name surname 0 bobby fisher 1 ronald fisher 2 ronald reagan 3 ronald mcdonald >>> df.columns Index([u'name', u'surname'], dtype='object') >>> df.index RangeIndex(start=0, stop=4, step=1)
As you can see:
- the keys of the dictionary became the
columnsofdf - the
indexof the variousSeriesbecame theindexofdf.
Warning
If the
indexof the inputSeriesdo not match, since label alignment applies, the missing values are treated asnan‘s.This is exactly what happens in this example:
>>> d = { >>> "name": >>> pd.Series([0, 0], index=["a", "b"]), >>> "surname": >>> pd.Series([0, 0], index=["c", "d"]), >>> } >>> df = pd.DataFrame(d) >>> df name surname a 0.0 NaN b 0.0 NaN c NaN 0.0 d NaN 0.0 >>> df.columns Index(['name', 'surname'], dtype='object') >>> df.index Index(['a', 'b', 'c', 'd'], dtype='object')
- the keys of the dictionary became the
Create a
DataFramefrom a single dictionary:>>> d = { ... "column1": [1., 2., 6., -1.], ... "column2": [0., 1., -2., 4.], ... } >>> df = pd.DataFrame(d) >>> df column1 column2 0 1.0 0.0 1 2.0 1.0 2 6.0 -2.0 3 -1.0 4.0
Again, the
columnsare taken from the keys, while theindexis set to the default one (i.e.range(0, num_rows)).A custom
indexcan be specified the usual way:>>> df = pd.DataFrame(d, index=["a", "b", "c", "d"]) >>> df column1 column2 a 1.0 0.0 b 2.0 1.0 c 6.0 -2.0 d -1.0 4.0
Create a
DataFramefrom a list of dictionaries:>>> data = [ ... {"a": 1, "b": 2}, ... {"a": 2, "c": 3}, ... ] >>> df = pd.DataFrame(data) >>> df a b c 0 1 2.0 NaN 1 2 NaN 3.0
Here the
columnsare taken (again) from the keys of the dictionaries, while theindexis the default one. Since not all common keys appear in all input dictionaries, missing values (i.e.nan‘s) are automatically added todf.
Note
Once a DataFrame has been created, you can change interactively its
index and columns:
>>> d = {
... "column1": [1., 2., 6., -1.],
... "column2": [0., 1., -2., 4.],
... }
>>> df = pd.DataFrame(d)
>>> df
column1 column2
0 1.0 0.0
1 2.0 1.0
2 6.0 -2.0
3 -1.0 4.0
>>> df.columns = ["bar", "bar"]
>>> df.columns
Index([u'foo', u'bar'], dtype='object')
>>> df.index = range(df.shape[1])
>>> df.index
Int64Index([0, 1, 2, 3], dtype='int64', length=4)
Loading a CSV file¶
Of course, the advantage of Pandas is that it allows to load data from many different file formats. Here we will only consider CSV (comma separated values) files and similar.
To load a CSV into a DataFrame, for instance the iris dataset, type:
>>> df = pd.read_csv("iris.csv")
>>> print type(df)
<class 'pandas.core.frame.DataFrame'>
Now that it’s loaded, let’s print some information about it:
>>> df.columns
Index([u'SepalLength', u'SepalWidth', u'PetalLength', u'PetalWidth', u'Name'], dtype='object')
>>> df.index
RangeIndex(start=0, stop=150, step=1)
>>> df.shape
(150, 5)
>>> print df
...
Note
The CSV file format is not really well-defined.
This means that the CSV files that you’ll encounter in the wild can (and probably will!) be very different from one another.
Given the diversity of this (under-specified) file format, the
read_csv() provides a large number of options to deal with many
of the (otherwise code-breaking) differences between CSV-like files.
Here is an excerpt from the read_csv() manual:
>>> help(pd.read_csv)
Help on function read_csv in module pandas.io.parsers:
read_csv(filepath_or_buffer, sep=',', delimiter=None, header='infer',
names=None, index_col=None, usecols=None, squeeze=False,
prefix=None, mangle_dupe_cols=True, dtype=None, engine=None,
converters=None, true_values=None, false_values=None,
skipinitialspace=False, skiprows=None, nrows=None, na_values=None,
keep_default_na=True, na_filter=True, verbose=False,
skip_blank_lines=True, parse_dates=False,
infer_datetime_format=False, keep_date_col=False, date_parser=None,
dayfirst=False, iterator=False, chunksize=None,
compression='infer', thousands=None, decimal='.',
lineterminator=None, quotechar='"', quoting=0, escapechar=None,
comment=None, encoding=None, dialect=None, tupleize_cols=False,
error_bad_lines=True, warn_bad_lines=True, skipfooter=0,
skip_footer=0, doublequote=True, delim_whitespace=False,
as_recarray=False, compact_ints=False, use_unsigned=False,
low_memory=True, buffer_lines=None, memory_map=False,
float_precision=None)
Read CSV (comma-separated) file into DataFrame
Also supports optionally iterating or breaking of the file
into chunks.
Additional help can be found in the `online docs for IO Tools
<http://pandas.pydata.org/pandas-docs/stable/io.html>`_.
As you can see, you can tweak the read_csv() method in very many
ways, in order to make it load your data the proper way.
Note
A similar function esists for reading Excel files, read_excel().
Example: A malformed TSV file. Let’s load a TAB-separated file instead (i.e. a CSV file that uses TABs instead of commas). The file can be found here:
It describes a mapping from UniProt protein IDs (i.e. sequences) to hits in the Protein Data Bank (i.e. structures). The TSV file looks like this:
# 2014/07/08 - 14:59
SP_PRIMARY PDB
A0A011 3vk5;3vka;3vkb;3vkc;3vkd
A0A2Y1 2jrd
A0A585 4mnq
A0A5B4 2ij0
A0A5B9 2bnq;2eyr;2eys;2eyt;3kxf;3o6f;3o8x;3o9w;3qux;3t0e;3ta3;3tvm
A0A5E3 1hq4;1ob1
A0AEF5 4iu2;4iu3
A0AEF6 4iu2;4iu3
A0AQQ7 2ib5
Note that the two columns are separated by a TAB. Note also that the first row is actually a comment.
We can use the sep (separator) argument of the read_csv() function
to take care of the TABs. Let’s write:
>>> df = pd.read_csv("uniprot_pdb.tsv", sep="\t")
>>> df.shape
(33637, 1)
>>> df.columns
Index([u'# 2014/07/08 - 14:59'], dtype='object')
>>> df.head()
# 2014/07/08 - 14:59
SP_PRIMARY PDB
A0A011 3vk5;3vka;3vkb;3vkc;3vkd
A0A2Y1 2jrd
A0A585 4mnq
A0A5B4 2ij0
>>> df.head().shape
(5, 1)
Unfortunately, the shape of the DataFrame is wrong: there is only one
column!
The problem is that the very first line (the comment) contains no tabs: this makes Panda think that there is only one column.
We have to tell read_csv() to either ignore the first line:
>>> df = pd.read_csv("uniprot_pdb.tsv", sep="\t", skiprows=1)
>>> df.shape
(33636, 2)
>>> df.head()
SP_PRIMARY PDB
0 A0A011 3vk5;3vka;3vkb;3vkc;3vkd
1 A0A2Y1 2jrd
2 A0A585 4mnq
3 A0A5B4 2ij0
4 A0A5B9 2bnq;2eyr;2eys;2eyt;3kxf;3o6f;3o8x;3o9w;3qux;3...
>>> df.shape
(33636, 2)
or, as an alternative, to ignore all comment lines, i.e. all lines that start
with a sharp "#" character:
>>> df = pd.read_csv("uniprot_pdb.tsv", sep="\t", comment="#")
>>> df.shape
(33636, 2)
>>> df.head()
SP_PRIMARY PDB
0 A0A011 3vk5;3vka;3vkb;3vkc;3vkd
1 A0A2Y1 2jrd
2 A0A585 4mnq
3 A0A5B4 2ij0
4 A0A5B9 2bnq;2eyr;2eys;2eyt;3kxf;3o6f;3o8x;3o9w;3qux;3...
>>> df.head().shape
(5, 2)
Now the number of columns is correct!
Example: microarray data. Let’s also try to load the microarray file in Stanford PCL format (namely, a disguised TAB-separated-values file) available here:
Again, it is pretty easy:
>>> df = pd.read_csv("2010.DNAdamage.pcl", sep="\t")
>>> df.shape
(6128, 55)
>>> df.columns
Index([u'YORF', u'NAME', u'GWEIGHT', u'wildtype + 0.02% MMS (5 min)',
u'wildtype + 0.02% MMS (15 min)', u'wildtype + 0.02% MMS (30 min)',
u'wildtype + 0.2% MMS (45 min)', u'wildtype + 0.02% MMS (60 min)',
u'wildtype + 0.02% MMS (90 min)', u'wildtype + 0.02% MMS (120 min)',
u'mec1 mutant + 0.02% MMS (5 min)', u'mec1 mutant + 0.02% MMS (15 min)',
u'mec1 mutant + 0.02% MMS (30 min)',
u'mec1 mutant + 0.02% MMS (45 min)',
u'mec1 mutant + 0.02% MMS (60 min)',
u'mec1 mutant + 0.02% MMS (90 min)',
u'mec1 mutant + 0.02% MMS (120 min)',
u'dun1 mutant + 0.02% MMS (30 min)',
u'dun1 mutant + 0.02% MMS (90 min)',
u'dun1 mutant + 0.02% MMS (120 min)',
u'wildtype + gamma irradiation (5 min)',
u'wildtype + gamma irradiation (10 min)',
...
u'crt1 mutant vs wild type (expt 1)',
u'crt1 mutant vs. wild type (expt 2)',
u'GAL-inducible ROX1 on galactose'],
dtype='object')
Warning
... at least at a quick glance.
Additional care must be taken for this data: some columns are not
separated correctly (just look at the file with the less command
to see what I mean).
Feel free to try and improve the example above to properly load the microarray file!
Extracting Rows and Columns¶
Row and column extraction from a DataFrame can be very easy to very
complex, depending on what you want to do.
Here we will only deal with elementary extraction methods.
Note
If you wish to know more, especially about the ever useful ix attribute,
take a look at the documentation:
The (non-exhaustive) list of alternatives is:
| Operation | Syntax | Result |
|---|---|---|
| Select column | df[col] |
Series |
| Select row by label | df.loc[label] |
Series |
| Select row by integer location | df.iloc[loc] |
Series |
| Slice rows | df[5:10] |
DataFrame |
| Select rows by boolean vector | df[bool_vec] |
DataFrame |
Note
For simplicity, in the following examples we will use a random sample taken from the iris dataset, computed like this:
>>> import numpy as np
>>> import pandas as pd
>>> np.random.seed(0)
>>> df = pd.read_csv("iris.csv")
>>> small = df.iloc[np.random.permutation(df.shape[0])].head()
>>> small.shape
(5, 5)
>>> small
SepalLength SepalWidth PetalLength PetalWidth Name
114 5.8 2.8 5.1 2.4 Iris-virginica
62 6.0 2.2 4.0 1.0 Iris-versicolor
33 5.5 4.2 1.4 0.2 Iris-setosa
107 7.3 2.9 6.3 1.8 Iris-virginica
7 5.0 3.4 1.5 0.2 Iris-setosa
Brief explanation: here we use numpy.random.permutation() to generate
a random permutation of the indices from 0 to df.shape[0], i.e.
the number of rows in the iris dataset; then we use these random
numbers as row indices to permute all the rows in df; finally, we
take the first 5 rows of the permuted df using the head()
method.
A
DataFramecan now be accessed like a regular dictionary to extract individual columns – indeed, you can also print its keys!:>>> small.columns Index([u'SepalLength', u'SepalWidth', u'PetalLength', u'PetalWidth', u'Name'], dtype='object') >>> small.keys() Index([u'SepalLength', u'SepalWidth', u'PetalLength', u'PetalWidth', u'Name'], dtype='object')
Now we access
smallby column name; the result is aSeries:>>> type(small["Name"]) <class 'pandas.core.series.Series'> >>> small["Name"] 114 Iris-virginica 62 Iris-versicolor 33 Iris-setosa 107 Iris-virginica 7 Iris-setosa Name: Name, dtype: object
If the name of the column is compatible with the Python conventions for variable names, you can also treat columns as if they were actual attributes of the
dfobject:>>> small.Name 114 Iris-virginica 62 Iris-versicolor 33 Iris-setosa 107 Iris-virginica 7 Iris-setosa Name: Name, dtype: object
Warning
Here
Nameis a variable created on-the-fly by Pandas when loading the iris setosa dataset!It is possible to extract multiple columns in one go; the result will be a
DataFrame:>>> small[["SepalLength", "PetalLength"]] SepalLength PetalLength 114 5.8 5.1 62 6.0 4.0 33 5.5 1.4 107 7.3 6.3 7 5.0 1.5
Warning
Note that the order of the column labels matters!
Compare this with the previous code fragment:
>>> small[["PetalLength", "SepalLength"]] PetalLength SepalLength 114 5.1 5.8 62 4.0 6.0 33 1.4 5.5 107 6.3 7.3 7 1.5 5.0
To extract the rows, we can use the
locandilocattributes:locallows to retrieve a row by label:>>> small.loc[114] SepalLength 5.8 SepalWidth 2.8 PetalLength 5.1 PetalWidth 2.4 Name Iris-virginica Name: 114, dtype: object >>> type(small.loc[114]) <class 'pandas.core.series.Series'>
The result is a
Series.ilocallows to retrieve a row by position:>>> small.iloc[0] SepalLength 5.8 SepalWidth 2.8 PetalLength 5.1 PetalWidth 2.4 Name Iris-virginica Name: 114, dtype: object
The result is, again, a
Series.The
locandilocattribute also allow to retrieve multiple rows:>>> small.loc[[114,7,62]] SepalLength SepalWidth PetalLength PetalWidth Name 114 5.8 2.8 5.1 2.4 Iris-virginica 7 5.0 3.4 1.5 0.2 Iris-setosa 62 6.0 2.2 4.0 1.0 Iris-versicolor >>> small.iloc[[0,1,2]] SepalLength SepalWidth PetalLength PetalWidth Name 114 5.8 2.8 5.1 2.4 Iris-virginica 62 6.0 2.2 4.0 1.0 Iris-versicolor 33 5.5 4.2 1.4 0.2 Iris-setosa
This time, the result is a
DataFrame.Warning
Again, note that the order of the row labels matters!
Broadcasting, Masking, Label Alignment¶
All of these work just like with Series:
Broacasting is applied automatically to all rows:
>>> small["SepalLength"] + small["SepalWidth"] 114 8.6 62 8.2 33 9.7 107 10.2 7 8.4 dtype: float64
or to the whole table:
>>> small + small SepalLength SepalWidth PetalLength PetalWidth \ 114 11.6 5.6 10.2 4.8 62 12.0 4.4 8.0 2.0 33 11.0 8.4 2.8 0.4 107 14.6 5.8 12.6 3.6 7 10.0 6.8 3.0 0.4 Name 114 Iris-virginicaIris-virginica 62 Iris-versicolorIris-versicolor 33 Iris-setosaIris-setosa 107 Iris-virginicaIris-virginica 7 Iris-setosaIris-setosa
The usual label alignment (and corresponding mismatching-labels-equals-
nan) rules apply:>>> small["SepalLength"][1:] + small["SepalWidth"][:-1] 7 NaN 33 9.7 62 8.2 107 10.2 114 NaN dtype: float64
Warning
Both row labels and column labels are automatically aligned!
Masking works as well, for instance:
>>> small.PetalLength[small.PetalLength > 5] 114 5.1 107 6.3 Name: PetalLength, dtype: float64 >>> small["PetalLength"][small["PetalLength"] > 5] 114 5.1 107 6.3 Name: PetalLength, dtype: float64 >>> small["Name"][small["PetalLength"] > 5] 114 Iris-virginica 107 Iris-virginica Name: Name, dtype: object >>> small[["Name", "PetalLength", "SepalLength"]][small.Name == "Iris-virginica"] Name PetalLength SepalLength 114 Iris-virginica 5.1 5.8 107 Iris-virginica 6.3 7.3
Statistics can be computed on rows:
>>> df.loc[114][:-1].mean() 4.0249999999999995
(Here the
[:-1]skips over theNamecolumn), on columns:>>> df.PetalLength.mean() 3.7586666666666662 >>> df["PetalLength"].mean() 3.7586666666666662
or on the whole table:
>>> df.mean() SepalLength 5.843333 SepalWidth 3.054000 PetalLength 3.758667 PetalWidth 1.198667 dtype: float64
or between rows:
>>> small.PetalLength.cov(small.SepalLength) 1.6084999999999996
You can combine them with masking as well:
>>> small[["Name", "PetalLength", "SepalLength"]][small.PetalLength > small.PetalLength.mean()] Name PetalLength SepalLength 114 Iris-virginica 5.1 5.8 62 Iris-versicolor 4.0 6.0 107 Iris-virginica 6.3 7.3
Merging Tables¶
Merging different dataframes is performed using the merge() function.
Merging means that, given two tables with a common column name, first the rows with the same column value are matched; then a new table is created by concatenating the matching rows.
Here is an explanatory image:
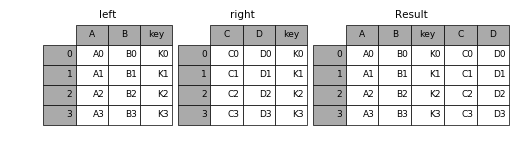
(Taken from the Merge, join, and concatenate section of the official Pandas documentation.)
Note
Only the column name must be the same in both tables: the actual values in the column may differ.
What happens when the column values are different, depends on the
how argument of the merge() function. See below.
A simple example of merging two tables describing protein properties (appropriately simplified for ease of exposition):
sequences = pd.DataFrame({
"id": ["Q99697", "O18400", "P78337", "Q9W5Z2"],
"seq": ["METNCR", "MDRSSA", "MDAFKG", "MTSMKD"],
})
names = pd.DataFrame({
"id": ["Q99697", "O18400", "P78337", "P59583"],
"name": ["PITX2_HUMAN", "PITX_DROME",
"PITX1_HUMAN", "WRK32_ARATH"],
})
print "input dataframes:"
print sequences
print names
print "full inner:"
print pd.merge(sequences, names, on="id", how="inner")
print "full left:"
print pd.merge(sequences, names, on="id", how="left")
print "full right:"
print pd.merge(sequences, names, on="id", how="right")
print "full outer:"
print pd.merge(sequences, names, on="id", how="outer")
Note that the "id" column appears in both dataframes. However, the values
in the column differ: "Q9W5Z2" only appears in the sequences table,
while "P59583" only appears in names.
Let’s see what happens using the four different joining semantics:
input dataframes:
id seq
0 Q99697 METNCR
1 O18400 MDRSSA
2 P78337 MDAFKG
3 Q9W5Z2 MTSMKD
id name
0 Q99697 PITX2_HUMAN
1 O18400 PITX_DROME
2 P78337 PITX1_HUMAN
3 P59583 WRK32_ARATH
# mismatched ids are dropped
full inner:
id seq name
0 Q99697 METNCR PITX2_HUMAN
1 O18400 MDRSSA PITX_DROME
2 P78337 MDAFKG PITX1_HUMAN
# ids are taken from the left table
full left:
id seq name
0 Q99697 METNCR PITX2_HUMAN
1 O18400 MDRSSA PITX_DROME
2 P78337 MDAFKG PITX1_HUMAN
3 Q9W5Z2 MTSMKD NaN
# ids are taken from the right table
full right:
id seq name
0 Q99697 METNCR PITX2_HUMAN
1 O18400 MDRSSA PITX_DROME
2 P78337 MDAFKG PITX1_HUMAN
3 P59583 NaN WRK32_ARATH
# all ids are retained
full outer:
id seq name
0 Q99697 METNCR PITX2_HUMAN
1 O18400 MDRSSA PITX_DROME
2 P78337 MDAFKG PITX1_HUMAN
3 Q9W5Z2 MTSMKD NaN
4 P59583 NaN WRK32_ARATH
To summarize, the how argument establishes the semantics for the mismatched
labels, and where the nan‘s should be inserted.
Example. Dataframe merging automatically takes care of replicating rows appropriately if the same key appears twice.
For instance, consider this code:
sequences = pd.DataFrame({
"id": ["Q99697", "Q99697"],
"seq": ["METNCR", "METNCR"],
})
names = pd.DataFrame({
"id": ["Q99697", "O18400", "P78337", "P59583"],
"name": ["PITX2_HUMAN", "PITX_DROME",
"PITX1_HUMAN", "WRK32_ARATH"],
})
print "input dataframes:"
print sequences
print names
print "full:"
print pd.merge(sequences, names, on="id", how="inner")
The result is:
input dataframes:
id seq
0 Q99697 METNCR
1 Q99697 METNCR
id name
0 Q99697 PITX2_HUMAN
1 O18400 PITX_DROME
2 P78337 PITX1_HUMAN
3 P59583 WRK32_ARATH
full:
id seq name
0 Q99697 METNCR PITX2_HUMAN
1 Q99697 METNCR PITX2_HUMAN
As you can see, rows are replicated appropriately!
Hint
This is extremely useful.
Consider for instance the problem of having to compute statistics on all the three gene-exon-CDS tables in the exercises section, jointly.
Using merge() it is easy to merge the three tables in a single, big
table, where the gene information is appropriately replicated for all
gene exons, and the exon information is replicated for all coding
sequences!
Note
The merge() method also allows to join tables by more than one column,
using the key attribute. See the official documentation here:
Grouping Tables¶
The groupby method is essential for efficiently performing operations on
groups of rows – especially split-apply-aggregate operations.
A picture is worth one thousand words, so here it is:
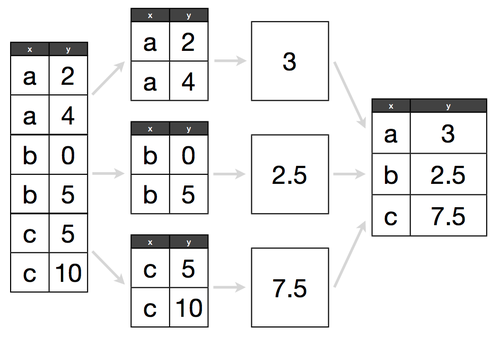
(Picture source: Hadley Wickham’s Data Science in R slides)
The groupby() method allows to extract groups of rows with the same value
(the “split” part), perform some computation on them (the “apply” part) and
then combine the results into a single table over the groups (the “aggregate”
part).
Example. Let’s take the iris dataset. We want to compute the average of the four data tables for the three different iris species.
The result should be a 3 species (rows) by 4 columns (petal/sepal length/width) dataframe.
First, let’s load the CSV file and print some information:
>>> iris = pd.read_csv("iris.csv")
>>> iris.info()
<class 'pandas.core.frame.DataFrame'>
RangeIndex: 150 entries, 0 to 149
Data columns (total 5 columns):
SepalLength 150 non-null float64
SepalWidth 150 non-null float64
PetalLength 150 non-null float64
PetalWidth 150 non-null float64
Name 150 non-null object
dtypes: float64(4), object(1)
memory usage: 5.9+ KB
Here is how to do that with groupby(). First, let’s group all rows of the
dataframe by the Name column:
>>> grouped = iris.groupby(iris.Name)
>>> type(grouped)
<class 'pandas.core.groupby.DataFrameGroupBy'>
>>> for group in grouped:
... print group[0], group[1].shape
...
Iris-setosa (50, 5)
Iris-versicolor (50, 5)
Iris-virginica (50, 5)
The groupby method returns a pandas.DataFrameGroupBy object (the
details are not important).
- To select a single group, write::
>>> group_df = grouped.get_group("Iris-versicolor") >>> print type(group) <class 'pandas.core.frame.DataFrame'> >>> group.info() Int64Index: 50 entries, 50 to 99 Data columns (total 5 columns): SepalLength 50 non-null float64 SepalWidth 50 non-null float64 PetalLength 50 non-null float64 PetalWidth 50 non-null float64 Name 50 non-null object dtypes: float64(4), object(1) memory usage: 2.3+ KB
Note
As shown by the above code snippet, iterating over the grouped variable
returns (value-of-Name, DataFrame) tuples:
- The first element is the value of the column
Nameshared by the group - The second element is a
DataFrameincluding only the rows in that group.
The whole idea is that you can apply some transformation (e.g. mean()) to
the individual groups automatically, using the aggregate() method
directly on the grouped variable.
For instance, let’s compute the mean of all columns for the three groups:
>>> iris_mean_by_name = grouped.aggregate(pd.DataFrame.mean)
>>> iris_mean_by_name.info()
<class 'pandas.core.frame.DataFrame'>
Index: 3 entries, Iris-setosa to Iris-virginica
Data columns (total 4 columns):
SepalLength 3 non-null float64
SepalWidth 3 non-null float64
PetalLength 3 non-null float64
PetalWidth 3 non-null float64
dtypes: float64(4)
memory usage: 120.0+ bytes
So the result of aggregate() is a dataframe. Here is what it looks like:
>>> iris_mean_by_name
SepalLength SepalWidth PetalLength PetalWidth
Name
Iris-setosa 5.006 3.418 1.464 0.244
Iris-versicolor 5.936 2.770 4.260 1.326
Iris-virginica 6.588 2.974 5.552 2.026
Warning
Note that the Name colum of the iris dataframe becomes the
index of the aggregated iris_mean_by_name dataframe: it is not a
proper column anymore!
The only actual columns are:
>>> iris_mean_by_name.columns
Index([u'SepalLength', u'SepalWidth', u'PetalLength', u'PetalWidth'], dtype='object')
Note
The groupby() method also allows to group by more than one column,
using the key attribute. See the official documentation here:
Pandas: Exercises¶
Hint
You can hone your skill with the (simple) exercises at:
Given the iris dataset:
How many rows does it contain? How many columns?
Compute the average petal length
Compute the average of all numerical columns
Extract the petal length outliers (i.e. those rows whose petal length is 50% longer than the average petal length)
Compute the standard deviation of all columns, for each iris species
Extract the petal length outliers (as above) for each iris species
Extract the group-wise petal length outliers, i.e. find the outliers (as above) for each iris species using
groupby(),aggregate(), andmerge().Hint
You can pass
as_index=Falsetogroupby()to keep theoncolumn as an actual column (rather than turn it into the index of the aggregated dataframe). Example:grouped = iris.groupby("Name", as_index=False)
Note
All the necessary files for the following exercises are here:
You will also need to count_values() method of the class Sequence:
>>> df = pd.DataFrame({
... "col1": ["a", "a", "d", "c"],
... "col2": ["x", "w", "d", "w"]
... })
...
>>> df
col1 col2
0 a x
1 a w
2 d d
3 c w
>>> df.col1.value_counts()
a 2
c 1
d 1
Name: col1, dtype: int64
>>> df.col2.value_counts()
w 2
d 1
x 1
Name: col2, dtype: int64
Note
Please refer to the following figure to refresh the biological concepts necessary to understand the following exercises.
The file
gene_table.txtcontains summary annotation on all human genes, based on the Ensembl annotation:For each gene, this file contains:
gene_name based on the HGNC nomenclature:
gene_biotype for example protein_coding, pseudogene, lincRNA, miRNA etc. See here for a more detailed description of the biotypes:
http://vega.sanger.ac.uk/info/about/gene_and_transcript_types.html
chromosome on which the gene is located
strand on which the gene is located
transcript_count the number of known isoforms of the gene
The incipit of the file
gene_name,gene_biotype,chromosome,strand,transcript_count TSPAN6,protein_coding,chrX,-,5 TNMD,protein_coding,chrX,+,2 DPM1,protein_coding,chr20,-,6 SCYL3,protein_coding,chr1,-,5 C1orf112,protein_coding,chr1,+,9 ...
Based on this file, write a program that:
- computes the number of genes annotated for the human genome
- computes the minimum, maximum, average and median number of known isoforms per gene (consider the transcript_count column as a series).
- plots a histogram of the number of known isoforms per gene
- computes the number of different biotypes
- computes, for each gene_biotype, the number of associated genes (a histogram), and prints the gene_biotype with the number of associated genes in decreasing order
- computes the number of different chromosomes
- computes, for each chromosome, the number of genes it contains (again, a histogram), and prints the chromosome with the corresponding number of genes in increasing order.
- computes, for each chromosome, the percentage of genes located on the
+strand - computes, for each biotype, the average number of transcripts associated to genes belonging to the biotype
The file
transcript_table.txtcontains summary annotation on all human transcripts, based on Ensembl annotation:For each transcript, this file contains:
transcript_name, composed of the gene name plus a numeric identifier
transcript_biotype for example protein_coding, retained_intron, nonsense_mediated_decay, etc., see here for a more detailed description of biotypes:
transcript_length the length of the transcript (without considering introns an poly A tail)
utr5_length the length of the 5’ UTR region (without considering introns)
cds_length the length of the CDS region (without considering introns)
utr3_length the length of the 3’ UTR region (without considering introns)
exon_count the number of exons of the transcript
canonical_flag a boolean indicating if the isoform is canonical (i.e. the reference isoform of the gene) or not. Each gene has only one canonical isoform.
The incipit of the file
transcript_name,transcript_biotype,transcript_length,utr5_length,cds_length,utr3_length,exon_count,canonical_flag ARF5-001,protein_coding,1103,154,543,406,6,T M6PR-001,protein_coding,2756,469,834,1453,7,T ESRRA-002,protein_coding,2215,171,1272,772,7,F FKBP4-001,protein_coding,3732,187,1380,2165,10,T CYP26B1-001,protein_coding,4732,204,1539,2989,6,T ...
Based on this file, write a program that:
- computes the number of transcripts annotated for the human genome
- computes the minimum, maximum, average and median length of human transcripts
- computes the minimum, maximum, average and median length of the CDS of human transcripts (excluding values equal to 0)
- computes the percentage of human transcripts with a CDS length that is a multiple of 3 (excluding values equal to 0)
- computes the minimum, maximum, average and median length of the UTRs of human transcripts (excluding values equal to 0)
- computes, for each transcript_biotype, the number of associated transcripts (a histogram), and prints the transcript_biotype with the number of associated transcripts in decreasing order
- computes, for each transcript_biotype, the average transcript length, and prints the results in increasing order
- computes, for protein_coding transcripts, the average length of the 5’UTR, CDS and 3’ UTR
- computes, for each transcript_biotype and considering only canonical transcripts, the average number of exons
The file
exon_table.txtcontains summary annotation on human exons associated with canonical transcripts, based on the Ensembl annotation:For each exon, this file contains:
- transcript_name, the name of the transcript
- exon_id, the id of the exon
- exon_rank, the rank of the exon inside the transcript (
1for the first exon,2for the second exon, etc.) - exon_chrom_start, the genomic coordinate corresponding to the start of the exon
- exon_chrom_end, the genomic coordinate corresponding to the end of the exon
The incipit of the file
transcript_name,exon_id,exon_rank,exon_chrom_start,exon_chrom_end ARF5-001,ENSE00001872691,1,127588345,127588565 ARF5-001,ENSE00003494180,2,127589083,127589163 ARF5-001,ENSE00003504066,3,127589485,127589594 ARF5-001,ENSE00003678978,4,127590066,127590137 ARF5-001,ENSE00003676786,5,127590963,127591088 ...
Based on this file, write a program that:
- computes the minimum, maximum, average and median length of human exons
- given the name of a transcript, returns an ordered list of its exons, plus the corresponding length
Load the gene-expression timeseries from:
Then:
- How many genes are there? How many time steps?
- Are there any measurements not assigned to a gene? If there are, fill in the missing gene names by using the genes appearing above the missing entries. (Take a look at the missing data handling section above.)
- Plot the expression of gene
"ZFX"using a line plot. - Plot the mean and variance of the expression of all genes using a line plot.
- Find out which genes are more expressed at time 0, 30m, 3h, 6h, and 12h.
- Find out, for each gene, which other gene is the other most correlated.
- Draw the matrix of pairwise gene correlations using
plt.imshow().
Write a Python function that mimics the semantics of
merge(): it takes two dataframes, matches the row labels, and concatenates the matching rows.The result should be a list of lists (or, optionally, a
DataFrame).It should follow
merge()‘show="inner"semantics, i.e. drop all rows that can not be matched.Hint
Use the
iterrows()method to iterate over the rows of aDataFrame.Example:
for row in df.iterrows(): # do something with the row (it is a series!)
Same as above, but for the
how="outer"semantics.
Pandas: Solutions¶
Solutions:
import pandas as pd import matplotlib.pyplot as plt df_iris = pd.read_csv("iris.csv")
Solution:
# number of rows and columns print df_iris.shape # only rows print len(df_iris.index) # only columns print len(df_iris.columns)
Solution:
# average petal length print df_iris.PetalLength.mean()
Solution:
# average of all numeric columns print df_iris.mean()
Solution:
# rows corresponding to petal length outliers print df_iris[df_iris.PetalLength > 1.5*df_iris.PetalLength.mean()]
Solution:
# group-wise standard deviation grouped = df_iris.groupby(df_iris.Name) iris_std_by_name = grouped.aggregate(pd.DataFrame.std) print iris_std_by_name # or in a single row print df_iris.groupby(df_iris.Name).aggregate(pd.DataFrame.std)
Solution:
# grouped outliers for name, dataframe in df_iris.groupby(df_iris.Name): print dataframe[dataframe.PetalLength > 1.5*df_iris.PetalLength.mean()]
Solution:
# WARNING: there are *no* group-wise outliers! # group-wise outliers for name, dataframe in df_iris.groupby(df_iris.Name): print dataframe[dataframe.PetalLength > 1.5*dataframe.PetalLength.mean()] # using groupby-aggregate-merge grouped = df_iris.groupby(df_iris.Name, as_index=False) petlen_mean_by_name = grouped.aggregate(pd.DataFrame.mean) merged = pd.merge(iris, petlen_mean_by_name, on="Name") # use `print merged.columns` to take a peek at the column names; the # column_x columns come from the left table (i.e. the original iris # DataFrame), the column_y ones come # from the right table (i.e. the # group-wise means) print merged[merged.PetalLength_x > 1.5 * merged.PetalLength_y]
Solutions:
import pandas as pd import matplotlib.pyplot as plt df_gene = pd.read_csv("gene_table.txt")
Solution:
# number of genes (there is exactly one per row) print df_gene.shape[0]
Solution:
# minimum, maximum, average and median number of isoforms per gene print df_gene.transcript_count.min(), \ df_gene.transcript_count.max(), \ df_gene.transcript_count.mean(), \ df_gene.transcript_count.median()
Solution:
# plot a histogram of the number of known isoforms per gene df_gene.transcript_count.plot(kind="hist",bins=100) plt.show()
Solution:
# computes, for each gene_biotype, the number of associated genes grouped = df_gene.groupby(df_gene.gene_biotype) grouped_number_by_biotype = grouped.aggregate(pd.DataFrame.count) print grouped_number_by_biotype
Solution:
rows = [] for biotype, subdf in df_gene.groupby(df_gene.gene_biotype): rows.append((len(subdf), biotype)) # alternative 1 rows.sort() rows.reverse() print rows # alternative 2, more advanced print sorted(rows, reverse=True)
Solution:
print len(df_gene.chromosome.unique()) # or equivalently (the result is a numpy array) print df_gene.shape[0]
Solution: almost identical to the solution of point 5.
Solution:
for chromosome, subdf in df_gene.groupby(df_gene.chromosome): num_plus = float(len(subdf[subdf.strand == "+"])) perc_plus = 100 * num_plus / len(subdf) print chromosome, perc_plus
Solution:
df_gene.groupby(df_gene.gene_biotype, as_index=False) \ .aggregate(pd.DataFrame.mean)[['gene_biotype', 'transcript_count']]
Solutions:
import pandas as pd import matplotlib.pyplot as plt df_tt = pd.read_csv("transcript_table.txt")
Solution:
print df_tt.shape[0]
Solution:
# minimum, maximum, average and median length of human transcripts print df_tt.transcript_length.min(), \ df_tt.transcript_length.max(), \ df_tt.transcript_length.mean(), \ df_tt.transcript_length.median()
Solution:
# minimum, maximum, average and median length of the CDS of human transcripts (excluding values equal to 0) print (df_tt.cds_length[df_tt.cds_length>0]).min(), \ (df_tt.cds_length[df_tt.cds_length>0]).max(), \ (df_tt.cds_length[df_tt.cds_length>0]).mean(), \ (df_tt.cds_length[df_tt.cds_length>0]).median()
Solution:
# computes the percentage of human transcripts with a CDS length that is a multiple of 3 (excluding values equal to 0) cds_number = df_tt.cds_length[df_tt.cds_length>0].shape[0] cds_multiple_number = df_tt.cds_length[df_tt.cds_length>0][df_tt.cds_length%3==0].shape[0] print (float(cds_multiple_number)/float(cds_number))*100 # Is the percentage equal to 100%? # How can you explain coding sequences whose length is not multiple of 3? (remember the rules of the genetic code)
Solution:
# minimum, maximum, average and median length of the UTRs of human transcripts (excluding values equal to 0) # for 5' UTRs filter_df = df_tt[df_tt.utr5_length>0] print filter_df.utr5_length.min(), \ filter_df.utr5_length.max(), \ filter_df.utr5_length.mean(), \ filter_df.utr5_length.median() # for 3' UTRs filter_df = df_tt[df_tt.utr3_length>0] print filter_df.utr3_length.min(), \ filter_df.utr3_length.max(), \ filter_df.utr3_length.mean(), \ filter_df.utr3_length.median() # for both 5' and 5' UTRs (calculating the sum of lengths) filter_df = df_tt[df_tt.utr5_length>0][df_tt.utr3_length>0] sum_of_lengths = filter_df.utr5_length + filter_df.utr3_length print sum_of_lengths.min(), \ sum_of_lengths.max(), \ sum_of_lengths.mean(), \ sum_of_lengths.median()
Solution:
# computes, for each transcript_biotype, the number of associated #transcripts (a histogram), and prints the transcript_biotype with the #number of associated transcripts in decreasing order grouped = df_tt.groupby(df_tt.transcript_biotype) grouped_number_by_biotype = grouped.size() # the result is a series grouped_number_by_biotype.sort(ascending=0) # sort the series print grouped_number_by_biotype
Solution:
# computes, for each transcript_biotype, the average transcript length, and prints the results in increasing order grouped = df_tt.groupby(df_tt.transcript_biotype) grouped_number_by_biotype = grouped.aggregate(pd.DataFrame.mean) sorted_number_by_biotype = grouped_number_by_biotype.sort("transcript_length") print sorted_number_by_biotype.transcript_length
Solution:
# computes, for protein_coding transcripts, the average length of the 5'UTR, CDS and 3' UTR filter_df = df_tt[df_tt.transcript_biotype=="protein_coding"] print filter_df.utr5_length.mean(), \ filter_df.cds_length.mean(), \ filter_df.utr3_length.mean()
Solution:
# computes, for each transcript_biotype and considering only canonical transcripts, the average number of exons filter_df = df_tt[df_tt.canonical_flag=="T"] grouped = filter_df.groupby(filter_df.transcript_biotype) grouped_number_by_biotype = grouped.aggregate(pd.DataFrame.mean) print grouped_number_by_biotype.exon_count
Solutions:
import pandas as pd import matplotlib.pyplot as plt df_exon = pd.read_csv("exon_table.txt")
Solution:
df_length = df_exon.exon_chrom_start - df_exon.exon_chrom_end + 1 print df_length.min(), df_length.max(), df_length.mean(), df_length.median()
Solution:
name = raw_input("write a trascript name :") filtered = df_exon[df_exon.transcript_name == name] filtered_length = filtered.exon_chrom_start - filtered.exon_chrom_end + 1 merged = df.merge(filtered, filtered_length) merged.sort(["exon_chrom_start"])
Solutions:
ts = pd.read_csv("expression_timeseries.txt", sep="\t")
Solution:
# number of genes print ts.shape[0] # number of time steps (just count how many columns end by 'h') print len([c for c in ts.columns if c[-1] == "h"])
Solution:
# if there are NaNs, then dropna will remove at least one row print "got nans?", ts.shape[0] != ts.dropna().shape[0] # how many rows with at least one NaN are there? (just to double check) print ts.shape[0] - ts.dropna().shape[0] # fill in the missing values ts = ts.ffill()
Note
In the following solutions we will assume that ffill() has already been called.
Solution: there are actually two expression profiles for the
"ZFX"gene:>>> expr_zfx = ts[ts["Gene Symbol"] == "ZFX"] >>> expr_zfx SPOT Gene Symbol 0h 0.5h 3h 6h 12h 0 1 ZFX -0.092 0.126 0.109 0.043 0.074 1739 1740 ZFX -0.487 0.298 -0.058 -0.158 -0.155
Let’s plot the first; the other can be dealt with similarly:
# first is a Series first = expr_zfx.iloc[0] numbers = first[2:] numbers.plot(kind="line") plt.savefig("output.png")
Solution: let’s take the mean over the time steps:
means = ts[ts.columns[2:]].mean(axis=1)
Here the first part extracts only those columns that encode expression measurements (from the third onwards), while
axis=1specifies that the average should be taken by averaging over columns, rather than over rows as we are used to. The latter case corresponds toaxis=0, and is the default.Now:
variances = ts[ts.columns[2:]].var(axis=1)
does the same thing for the variance.
Let’s plot the mean and the variance:
means.plot(kind="line") plt.savefig("means.png") # clear the screen between plots plt.clf() # same thing for variance here
Warning
The above exercise is slightly bugged. In order to be solved “the Pandas way”, it requires more advanced material that is not provided by the notes. I gave a fully Pandas-approved solution.
Solution:
# we know that the only columns we care about are from the third # onwards -- for simplicity for column in ts.columns[2:]: index_of_max = ts[column].argmax() print column, ts.iloc[index_of_max]
Unfortunately most of the gene names are
NaN; the solution is correct nevertheless.Solution: same as below, except that the inner loop keeps track of the index of the maximally correlated gene.
Very elementary solution, it will take a long time to complete:
ts = pd.read_csv("expression_timeseries.txt", sep="\t") num_rows = ts.shape[0] # set this to 100 or so to make it faster corr = [] for i in range(num_rows): row_i = ts.iloc[i][2:].astype(float) corr_i = [] for j in range(num_rows): row_j = ts.iloc[j][2:].astype(float) corr_i.append(row_i.corr(row_j)) corr.append(corr_i) plt.imshow(pd.DataFrame(corr).values, cmap="gray", interpolation="nearest") plt.savefig("corr.png")
Left to the reader.
Numpy: a cursory overview¶
Warning
This is an appendix.
This material will not be part of the final exam.
Numpy is a freely available library for performing efficient numerical computations over scalars, vectors, matrices, and more generally N-dimensional tensors.
Its main features are:
- Flexible indexing and manipulation of arbitrary dimensional data.
- Many numerical routines (linear algebra, Fourier analysis, local and global optimization, etc.) using a variety of algorithms.
- Clean Python interface to many underlying libraries (for instance, the GNU Scientific Library, or GSL), which are used as efficient implementations of numerical algorithms.
- Support for random sampling from a variety of distributions.
This list is far from complete!
Documentation & Source¶
The official numpy website is:
The numpy source code is available at:
and is distributed under the numpy license:
Importing numpy¶
The customary idiom to import numpy is:
>>> import numpy as np
>>> help(np)
Importing specific sub-modules also makes sense, so feel free to write (in this case for the linear algebra module):
>>> import numpy.linalg as la
>>> help(la)
The Array class¶
An ndarray is what it says: an N-dimensional array (also known as a
tensor).
The usual concepts encountered in analysis and algebra can be implemented as arrays: vectors are 1D arrays, matrices are 2D arrays.
Given an array x, its dimensionality and type can be ascertained by
using the shape, ndim and dtype attributes:
>>> x = np.zeros(10)
array([ 0., 0., 0., 0., 0., 0., 0., 0., 0., 0.])
>>> print type(x)
<type 'numpy.ndarray'>
>>> print x.shape
(10,)
>>> print x.ndim
1
>>> print x.dtype
float64
Creating an Array¶
Creating arrays from scratch can be useful to start off your mathematical code or for debugging purposes. A few useful creation functions are:
Creating an all-zero array is simple with
np.zeros():>>> # One dimensional case >>> x = np.zeros(2) >>> x array([ 0., 0.]) >>> # Two dimensional case >>> x = np.zeros((2, 2)) >>> x array([[ 0., 0.], [ 0., 0.]]) >>> x.shape (2, 2) >>> # Three dimensional case >>> x = np.zeros((2, 2, 2)) >>> x array([[[ 0., 0.], [ 0., 0.]], [[ 0., 0.], [ 0., 0.]]]) >>> x.shape (2, 2, 2)
Creating an all-ones array is equally simple with
np.ones():>>> # N-dimensional case >>> x = np.ones((2, 5, 5, 10)) >>> x.shape (2, 5, 5, 10)
To create a diagonal matrix, use either the
diag()andones()methods together, or theeye()method (reminiscent of Matlab):>>> np.diag(np.ones(3)) array([[ 1., 0., 0.], [ 0., 1., 0.], [ 0., 0., 1.]]) >>> np.eye(3) array([[ 1., 0., 0.], [ 0., 1., 0.], [ 0., 0., 1.]])
The numpy analogue of
range()isnp.arange():>>> x = np.arange(10) >>> x.shape (10,) >>> x array([0, 1, 2, 3, 4, 5, 6, 7, 8, 9])
Converting from an existing list or list-of-lists object:
>>> l = [range(3*i,3*(i+3),3) for i in range(3)] >>> l [[0, 3, 6], [3, 6, 9], [6, 9, 12]] >>> np.array(l) array([[ 0, 3, 6], [ 3, 6, 9], [ 6, 9, 12]])
In this case, the
dtypewill be based on the type of the elements in the inputlist.Random sampling an array from a distribution:
>>> # Sampling a single scalar from a real-valued uniform (sampling >>> # functions return a scalar if no shape information is given) >>> x = np.random.uniform(0, 10) >>> x 3.9300397597884897 >>> # Sampling a 3-element vector from a Normal distribution >>> # with mean=1 and sigma=0.2 >>> x = np.random.normal(1, 0.2, size=3) >>> x array([ 1.2001157 , 0.94376082, 1.09514026]) >>> # Sampling a binary 3x3 matrix, i.e. from a uniform >>> # distribution over integers >>> x = np.random.randint(0, 2, size=(3, 3)) >>> x array([[1, 0, 0], [1, 0, 1], [0, 1, 0]])
Example. To sample a random 100x100 matrix where every entry is distributed according to a normal distribution, write:
import numpy as np
import matplotlib.pyplot as plt
np.random.seed(0)
rm = np.random.normal(0, 1, size=(100, 100))
plt.imshow(rm, cmap="gray", interpolation="nearest")
plt.savefig("random-matrix.png")
The result is:
Note
The reference manual of the numpy.random module is here:
For a quick-and-dirty explanation of (pseudo!) random number generation as used in Computer Science, see:
The numpy.random RNG makes use of the very popular Mersenne Twister (MT)
PRNG. For an introduction, see here:
Warning
For reproducibility, make sure to initialize the pseudo-random number
generator with np.random.seed() before running your code, i.e.:
>>> np.random.seed(0)
After setting the seed, your code will be deterministic as long as calls to the sampling routines above remain the same.
From pandas to numpy and back¶
It is very easy to “convert” a Series or DataFrame into an array: as a
matter of facts, these Pandas types are built on numpy arrays: their
underlying array is accessible through the .values attribute.
(Pandas of course coats the underlying array with a lot of useful functionality!)
For instance, given a DataFrame of the iris dataset:
iris = pd.read_csv("iris.csv")
the underlying array can be accessed through its .values attribute:
>>> # For a table of two columns
>>> type(iris[["PetalLength", "SepalLength"]])
<class 'pandas.core.frame.DataFrame'>
>>> type(iris[["PetalLength", "SepalLength"]].values)
<type 'numpy.ndarray'>
The same can be done with individual Series objects, e.g. the columns
of the DataFrame:
>>> type(iris.PetalWidth.values)
<type 'numpy.ndarray'>
It is equally easy to construct a DataFrame out of an arrray: just call
the DataFrame passing it the array as an argument:
>>> pd.DataFrame(x)
0 1 2
0 0 1 2
1 3 4 5
2 6 7 8
If you also want to assign the column and row labels, you can pass them as optional arguments:
>>> pd.DataFrame(x,
... columns=['a', 'b', 'c'],
... index=['one', 'two', 'three'])
a b c
one 0 1 2
two 3 4 5
three 6 7 8
Reshaping¶
Reshaping. The arrangement of entries into rows, columns, tubes, etc. can be
changed at any time using the reshape() and ravel() methods:
>>> # Turn a 9-element vector into a 3x3 matrix
>>> x = np.arange(9)
>>> x
array([0, 1, 2, 3, 4, 5, 6, 7, 8])
>>> x.reshape((3, 3))
array([[0, 1, 2],
[3, 4, 5],
[6, 7, 8]])
>>> # Turn a 3x3 matrix into a 9-element vector
>>> x = np.ones((3, 3))
>>> x
array([[ 1., 1., 1.],
[ 1., 1., 1.],
[ 1., 1., 1.]])
>>> x.ravel()
array([ 1., 1., 1., 1., 1., 1., 1., 1., 1.])
Indexing¶
Given an array of shape (n1, n2, ..., nd), you can extract a single element
through its index.
For instance, in the 2D case:
>>> x = np.arange(9).reshape((3, 3))
>>> x
array([[0, 1, 2],
[3, 4, 5],
[6, 7, 8]])
>>> x[0,0] # first row, first column
0
>>> x[1,2] # second row, third column
5
>>> x[2,0] # thrid row, first column
6
In the example above, x[i,j] extracts the element in row i, column
j – just like in standard matrix algebra.
You can also use the colon : notation to select specify all the elements
on an axis, for instance a whole column, a whole row, or an entire sub-array:
>>> # The first row
>>> x[0,:]
array([0, 1, 2])
>>> # A shorthand for the first row
>>> x[0]
array([0, 1, 2])
>>> # The first column (no shorthand available)
>>> x[:,1]
array([1, 4, 7])
>>> # A sub-table
>>> x[1:3, :]
array([[3, 4, 5],
[6, 7, 8]])
The very same syntax applies to the general N-dimensional case:
>>> x = np.arange(10**5).reshape((10,)*5)
>>> x.shape
(10, 10, 10, 10, 10)
>>> x[1,2,3,4,5]
12345
>>> x[0,0,:,:,0]
array([[ 0, 10, 20, 30, 40, 50, 60, 70, 80, 90],
[100, 110, 120, 130, 140, 150, 160, 170, 180, 190],
[200, 210, 220, 230, 240, 250, 260, 270, 280, 290],
[300, 310, 320, 330, 340, 350, 360, 370, 380, 390],
[400, 410, 420, 430, 440, 450, 460, 470, 480, 490],
[500, 510, 520, 530, 540, 550, 560, 570, 580, 590],
[600, 610, 620, 630, 640, 650, 660, 670, 680, 690],
[700, 710, 720, 730, 740, 750, 760, 770, 780, 790],
[800, 810, 820, 830, 840, 850, 860, 870, 880, 890],
[900, 910, 920, 930, 940, 950, 960, 970, 980, 990]])
Note
It can be difficult to visualize operations over N-dimensional arrays, when N is larger than 3.
However, it is much easier when the axes have a concrete semantics. For
instance, assume that the 4D array performace holds the performance
of several sequence alignment algorithms over multiple sequence clusters
in multiple databases:
- The first axis is the index of the algorithm
- The second axis is the index of the database
- The third axis is the index of a cluster of sequences in the database
- The fourth axis is one of three measures of performance: precision, recall, and accuracy
Your tensor looks like this:
performances[alg][db][cluster][measure]
Then in extract the accuracy of algorithm 1 over all databases
and sequence clusters, you can just do:
performances[1,:,:,0]
or even:
performances[1,:,:,-1]
Now that the axes have a concrete semantics, it is much easier to see what this operation does!
Arithmetic and Functions¶
Operations between scalars and arrays are broadcast, like in Pandas (and more generally in linear algebra). For instance:
>>> x = np.arange(5)
>>> 10 * x
array([ 0, 10, 20, 30, 40])
>>> y = -x
>>> y
array([ 0, -10, -20, -30, -40])
Operations between equally-shaped arrays operate element-wise:
>>> x
array([0, 1, 2, 3, 4])
>>> x + x
array([0, 2, 4, 6, 8])
>>> x + y
array([0, 0, 0, 0, 0])
Operations can update the sub-arrays automatically:
>>> x = np.arange(16).reshape((4, 4))
>>> x
array([[ 0, 1, 2, 3],
[ 4, 5, 6, 7],
[ 8, 9, 10, 11],
[12, 13, 14, 15]])
>>> x[1:3,1:3] += 100 # Note the automatic assignment!
>>> x
array([[ 0, 1, 2, 3],
[ 4, 105, 106, 7],
[ 8, 109, 110, 11],
[ 12, 13, 14, 15]])
Operations between arrays of different shapes but compatible sizes are
broadcast by “matching” the last (right-most) dimension. For instance,
addition between a matrix x and a vector y broadcasts y over all
rows of x:
>>> x = np.arange(9).reshape((3, 3))
>>> y = np.arange(3)
>>> x + y
array([[ 0, 2, 4],
[ 3, 5, 7],
[ 6, 8, 10]])
The result, x + y, can be seen as the result of:
result = np.zeros(x.shape)
for i in range(x.shape[0]):
result[i,:] = x[i,:] + y[:]
As you can see, the last (right-most) dimension of x is matched to the last
(and only) dimension of y.
Warning
Operations between arrays of incompatible sizes raise an error:
>>> np.arange(3) + np.arange(2)
Traceback (most recent call last):
File "<stdin>", line 1, in <module>
ValueError: operands could not be broadcast together with shapes (3,) (2,)
Mathematical Functions¶
Most of the usual mathematical functions are implemented in numpy, including the usual max and min operators and trigonometric functions.
For instance:
>>> np.sin(0), np.cos(0)
(0.0, 1.0)
>>> np.sin(np.pi / 2), np.cos(np.pi / 2)
(1.0, 6.123233995736766e-17)
As you can see, the value of the cosine at \(\frac{\pi}{2}\) should be zero, but is approximated by a very (very) small value. The same is true for other functions:
>>> np.tan(np.pi / 4)
0.99999999999999989
The nice property of mathematical functions in numpy is that they are automatically applied to all elements of an array. For instance:
>>> x = [0, 0.25 * np.pi, 0.5 * np.pi, 0.75 * np.pi]
>>> np.cos(x)
array([ 1.00000000e+00, 7.07106781e-01, 6.12323400e-17,
-7.07106781e-01])
where the second and last elements are half the square root of two and its opposite:
>>> np.sqrt(2) / 2
0.70710678118654757
Complex mathematical expressions can be therefore specified very compactly using this property. For instance, in order to plot a sinewave with small additive normally-distributed noise, we can write:
import numpy as np
import matplotlib.pyplot as plt
# The x values
x = np.arange(100)
# The y values, uncorrupted
y = np.sin(2 * np.pi * np.arange(100) / 100.0)
plt.plot(x, y)
# The y values, now corrupted by Gaussian noise
y += np.random.normal(0, 0.05, size=100)
plt.plot(x, y)
plt.savefig("sin.png")
The result is:
Note
For the complete list of the available mathematical functions, see:
Linear Algebra¶
Numpy provides a number of linear algebraic functions, including dot products, matrix-vector multiplication, and matrix-matrix multiplication:
import numpy as np
# dot (scalar) product between vectors
x = np.array([0, 0, 1, 1])
print np.dot(x, x)
z = np.array([1, 1, 0, 0])
print np.dot(z, z)
print np.dot(x, z)
# Matrix-vector product
a = 2 * np.diag(np.ones(4))
print(np.dot(a, x))
print(np.dot(a, z))
# Matrix-matrix product
b = np.ones((4, 4)) - a
print(np.dot(a, b))
The result is:
# np.dot(x, x)
2
# np.dot(z, z)
2
# np.dot(x, z)
0
# np.dot(a, x)
[ 0. 0. 2. 2.]
# np.dot(a, z)
[ 2. 2. 0. 0.]
# np.dot(a, b)
[[-2. 2. 2. 2.]
[ 2. -2. 2. 2.]
[ 2. 2. -2. 2.]
[ 2. 2. 2. -2.]]
Numpy also provides routines for dealing with eigenvalues/eigenvects, singular value decompositions, and other decompositions.
Let’s compute the (trivial) eigenvalues/eigenvectors of the 4x4 identity matrix:
>>> import numpy.linalg as la
>>> eigenvalues, eigenvectors = la.eig(np.ones((4, 4)))
>>> eigenvalues
array([ 0., 4., 0., 0.])
>>> eigenvectors
array([[ -8.66025404e-01, 5.00000000e-01, -2.77555756e-17,
-2.77555756e-17],
[ 2.88675135e-01, 5.00000000e-01, -5.77350269e-01,
-5.77350269e-01],
[ 2.88675135e-01, 5.00000000e-01, 7.88675135e-01,
-2.11324865e-01],
[ 2.88675135e-01, 5.00000000e-01, -2.11324865e-01,
7.88675135e-01]])
Note
The reference manual of the numpy.linalg module is here:
numpy: Exercises¶
Implement a function that takes a
ddimensional vectorxreturns their Euclidean norm.Hint
The Euclidean norm of a vector is the square root of its dot product with itself.
Implement a function that takes two
ddimensional vectorsxandzand returns their Euclidean distance.Hint
Can you re-use the solution of the previous exercise?
Implement a function that takes a matrix
Aand an integerk, and returnsAelevated to thekth power.Write a Python program that plots the data here:
https://drive.google.com/open?id=0B0wILN942aEVVlk4TS1WaDItVU0
Every row has an experiment ID and a value; there are 10 experiments, and 100 values (rows) per experiment. For each experiment, plot its values as a time series.
The plot the average time series, i.e. the average curve, where the average is taken over all experiments.
Hint
Use
matplotlib.pyplotand theplot(x, y)function, as done in the examples above, to plot the ten curves and their mean.Implement the Power Iteration method for finding the largest eigenvalue and eigenvector of a matrix, as described in the first paragraph of:
Check that it matches the results given by the
eig()method of thelinalgmodule.Implement the Gram-Schmidt orthogonalization routine, described here:
Given the iris dataset, compute the covariance matrix of the petal lenght and petal widht for the iris setosa rows.
- Running a Linux Distro
- Unix shell: a few tips
- Python: Basics
- Python: Numbers
- Python: Numbers (Solutions)
- Python: Strings
- Python: Strings (Solutions)
- Python: Lists
- Python: Lists (Solutions)
- Python: Tuple
- Exercises
- Python: Tuple (Solutions)
- Python: Dictionaries
- Exercises
- Python: Dictionaries (Solutions)
- Python: Input-Output
- Exercises (raw input)
- Exercises (Filesystem)
- Python: Input-Output (Solutions)
- Python: Complex Statements
- Exercises:
if - Exercises:
forandwhile - Exercises: nested statements
- Python: complex statements (Solutions)
- Python: Functions
- Exercises
- Exercises
- Python: Functions (Solutions)
- Python: Programs
- Python: Programs (Solutions)
- External Modules
- Biopython
- Biopython (Exercises)
- Biopython (Solutions)
- Pandas
- What is Pandas?
- Documentation & Source
- Importing Pandas
- Pandas: A short demonstration
- Pandas datatypes
- Pandas:
Series - Creating a
Series - Accessing a
Series - Operator Broadcasting
- Masks and Filtering
- Automatic Label Alignment
- Dealing with Missing Data
- Computing Statistics
- Plotting
- Pandas:
DataFrame - Creating a
DataFrame - Loading a CSV file
- Extracting Rows and Columns
- Broadcasting, Masking, Label Alignment
- Merging Tables
- Grouping Tables
- Pandas: Exercises
- Pandas: Solutions
- Numpy: a cursory overview
- numpy: Exercises